
基质固相分散-高效液相色谱法测定川味香肠中的苏丹红染料
2012-10-26 00:42杨春林李佳峻彭灯水
食品工业科技 2012年9期
杨春林,李佳峻,胡 强,吴 蔚,蒙 辉,彭灯水
(1.乐山市产品质量监督检验所食品化工中心,四川乐山 614000;2.上海海丰现代农业有限公司,上海202179;3.四川农业大学资源环境学院,四川雅安 625014)
基质固相分散-高效液相色谱法测定川味香肠中的苏丹红染料
杨春林1,李佳峻1,胡 强1,吴 蔚1,蒙 辉2,彭灯水3
(1.乐山市产品质量监督检验所食品化工中心,四川乐山 614000;2.上海海丰现代农业有限公司,上海202179;3.四川农业大学资源环境学院,四川雅安 625014)
建立了川味香肠中苏丹红I~IV的基质固相分散(MSPD)-高效液相色谱(HPLC)分析方法。样品以无水硫酸钠作为分散基体,研磨均匀后与中性氧化铝同时装柱,正己烷淋洗净化,以丙酮-正己烷(5∶95,V/V)溶液洗脱。用Inertsil ODS-sp C18柱(4.6mm×250mm,5μm)进行分离,流动相A为含0.1%甲酸的甲醇,流动相B为0.02mol/L的乙酸铵溶液(A∶B= 85∶15,V/V),等度洗脱,柱温40℃。二极管阵列检测器检测,检测波长为490nm,利用保留时间和光谱图定性,外标法定量。4种苏丹红染料在0.10~50.00μg/mL范围内线性关系良好,相关系数均大于0.9999,苏丹红I、II、III、IV的检出限(LOD)分别为0.008~0.011mg/kg(信噪比S/N=3)。在添加浓度为5.0~25.0mg/kg范围内平均回收率达85.54%~94.66%,相对标准偏差(RSD)为0.87%~4.23%(n=6)。
川味香肠,苏丹红I~IV,高效液相色谱,基质固相分散
苏丹红(Sudan Dyes)属于偶氮类化工染色剂,主要用于为溶剂、油、蜡、汽油增色以及鞋、地板等的增光以及生化毒理学研究中的着色等[1-3]。据英国联邦危险品管理局测试,长期低量摄入苏丹红可能致癌[4]。研究表明,苏丹红本身不会对人体产生有害的影响,但它可以被还原降解为对人体具有强烈的致癌作用的芳香胺类化合物[5]。国际癌症研究机构(IARC)将苏丹红归为三类致癌物,包括我国在内的许多国家都明令禁止将其作为色素添加剂在食品和饲料中使用[6]。由于苏丹红颜色鲜艳、不易褪色并具有油溶性的特点,不法分子使用含苏丹红的辣椒作为川味香肠的调味料或直接用苏丹红代替红曲红天然色素给香肠上色,以降低生产成本,获得高额利润。所以建立有效的检测川味香肠中苏丹红的方法具有重要的意义。目前,食品中苏丹红染料的检测方法主要有高效液相色谱法[7-10]、液质联用法[11-12]、气质联用法[13]、分光光度法[14]、荧光光谱法[15]、共振光散射法[16]以及酶联免疫检测法等[17],常用的前处理技术主要包括溶剂萃取法(SE)、固相萃取法(SPE)、固相微萃取技术(SPME)等。我国颁布的标准方法[18]采用正己烷提取,中性氧化铝柱净化,该方法对辣椒面等含油脂和水分较少的样品提取和净化效果较好,回收率和灵敏度很高。基质固相分散技术(MSPD)[19]集合了传统的样品均质、提取、过滤、净化等过程,使样品的前处理简便快捷,减少有机溶剂的使用量,大大提高传统提取方法不易分散的样品的回收率,该技术已广泛用于动物组织和水果、蔬菜中农药残留的分析[20]。川味香肠含油脂和动物蛋白较多,不易分散,实验采用基质固相分散—氧化铝层析法来提取川味香肠中的苏丹红,用高效液相色谱—二极管阵列检测器进行检测,并优化前处理技术和色谱条件,建立川味香肠中苏丹红I~IV的基质固相分散—高效液相色谱测定方法。
1 材料与方法
1.1 材料与仪器
苏丹红Ⅰ、Ⅱ、Ⅲ、Ⅳ标准品 德国Dr.Ehrenstorfer公司;乙腈、甲醇、丙酮、正己烷(色谱纯) 美国Tedia公司;乙醚、甲酸、乙酸铵、无水硫酸钠(500℃烘烤3h) 成都市科龙化工试剂厂;层析用中性氧化铝(100~200目) 上海陆都化学试剂厂。
高效液相色谱仪LC-20A(配置四元泵溶剂淋洗系统、自动进样系统、光电二极管阵列检测器) 日本SHIMADZU公司;氮吹仪N-EVAP 美国Organomation公司;电子天平DV215CD(精确至0.00001g) 美国Ohaus公司;纯水机 四川沃特尔科技有限公司;超声波清洗器 上海冠特超声仪器有限公司。
1.2 实验方法
1.2.1 色谱条件 色谱柱:Inertsil ODS-sp C18(4.6mm× 250mm,5μm);流动相:含0.1%甲酸的甲醇+0.02mol/L的乙酸铵溶液(85∶15,V/V),使用前过0.45μm滤膜;柱温:40℃,流速:1.0m L/m in,等度洗脱;检测波长为490nm。
1.2.2 中性氧化铝制备 称取在200℃下烘烤2h的中性氧化铝100g,加入1.8m L水,混匀后密封备用。
1.2.3 标准曲线
1.2.3.1 标准储备液配制 准确称取苏丹红I、II、III、IV标准品各10mg(精确至0.01mg),用乙醚溶解并用丙酮稀释成浓度为1mg/m L的储备液。
1.2.3.2 标准曲线的建立 取标准储备液适量用丙酮稀释定容成浓度为0.10、0.50、1.25、5.00、10.00、25.00、 50.00μg/m L的工作溶液,取20μL进样测定峰面积,以质量浓度对峰面积作曲线。
1.2.4 样品处理及测定 取有代表性的川味香肠切细后匀浆,准确称取5g试样于100m L研钵中,加入5g无水硫酸钠,研磨均匀备用。取20cm×2cm玻璃层析柱,在其底部垫一层薄薄的脱脂棉,以干法装入制备好的中性氧化铝至约3cm高,轻轻敲击使填实后用10m L正己烷预淋洗;加入研磨好的均匀样品,轻轻敲实后于上部塞一层薄薄的脱脂棉,架于铁架台上。用20~30m L正己烷淋洗至无色,弃淋洗液,用50m L含5%丙酮的正己烷洗脱,收集洗脱液于塑料离心管中,氮吹仪浓缩至近干,用丙酮准确定容至5m L,0.45μm有机滤膜过滤后进行液相色谱分析,以保留时间结合光谱图定性,峰面积定量。
2 结果与分析
2.1 检测波长的选择
将苏丹红I~IV号标准混合溶液进行色谱分离并用二极管阵列检测器在波长200~550nm范围内进行紫外扫描,光谱图见图1。结果表明,在该方法仪器条件下四种苏丹红染料均在480~520nm波长范围内有最大吸收,其中苏丹红I和苏丹红II的最大吸收波长小于500nm,分别为476nm和495nm,苏丹红III和苏丹红IV的最大吸收波长大于500nm,为506nm和516nm。国家标准方法(GB/T19681-2005《食品中苏丹红染料的检测方法—高效液相色谱法》)采用的检测波长为500nm,在该波长条件下,苏丹红I~IV号均有较高的响应值,但是川味香肠实际样品的测定中发现,在500nm波长下苏丹红III和苏丹红IV色谱峰之间的干扰较多,通过筛选验证,选择490nm为苏丹红I~IV号的检测波长。

图1 苏丹红I~IV紫外可见扫描光谱图Fig.1 UV-visible absorption spectra of Sudan I~IV
2.2 色谱条件的选择和优化
在国家标准方法中,采用溶剂A:0.1%甲酸的水溶液∶乙腈=85∶15,溶剂B:0.1%甲酸的乙腈溶液∶丙酮=80∶20作为流动相,梯度洗脱。该方法大量使用乙腈、丙酮等高毒试剂,危害操作人员的身体健康,对环境很不友好。并且由于该方法采用梯度洗脱,在第10min有机相从75%改变至100%,为了平衡基线以进行下一个样品的分析,第32m in开始有机相又从100%变换到75%,所以分析过程的后半段基线的波动较大(图2A),对苏丹红III特别是苏丹红IV的准确定量造成一定的困难,并且从一个样品开始分析到下一个样品进样需要50m in甚至更长的时间。实验分别采用乙腈-水、甲醇-水、乙腈-甲酸水溶液、乙腈-硫酸铵水溶液和甲醇-硫酸铵水溶液作为流动相做等度洗脱,均不能将四种苏丹红完全分离;用酸性甲醇-水溶液能将目标物质分离,但是苏丹红IV色谱峰拖尾严重,在水溶液中加入0.02mmol/L的乙酸铵等度洗脱不但分离完全,并且峰型尖锐对称(图2B)。本方法采用等度洗脱将4种苏丹红完全分离,与国标方法相比四个目标物的出峰时间稍有提前,并且由于基线平稳,进样后25m in即可进行下一个样品的分析,大大缩短分析时间,提高工作效率,定量更加准确。
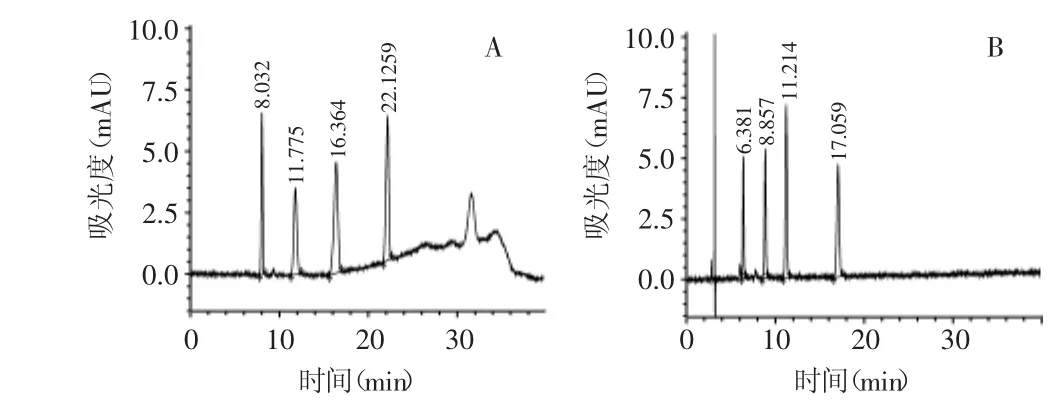
图2 苏丹红I~IV标准溶液色谱图Fig.2 Chromatogram of standard solution of Sudan I~IV
2.3 样品前处理优化及测定
2.3.1 基质固相分散—氧化铝层析法 香肠中含有大量的油脂和动物蛋白,而苏丹红又是油溶性的物质,在提取过程中由于样品中一部分动物蛋白变性后与油脂一起作用,将苏丹红包裹起来,用有机溶剂直接提取不能有效分散样品,提取率较低。按照国家标准方法用正己烷从香肠中提取苏丹红,浓缩后过中性氧化铝柱,由于大量油脂的集中过柱,影响中性氧化铝对苏丹红的吸附,加标回收率很低,其中苏丹红III的平均加标回收率只有40%左右。本实验针对肉制品的特点,采用基质固相分散—氧化铝层析法,用无水硫酸钠与样品一起研磨后再与中性氧化铝一起装柱,达到分散样品的目的,使提取溶剂与样品充分接触。由于样品没有浓缩,油脂分散进入氧化铝柱,对氧化铝活性影响较小;加入无水硫酸钠还可以消除香肠中水分对氧化铝吸附活性的影响,提高样品回收率。
自制的层析柱的分离效果受多方面因素的影响,对操作技术的要求较高,在装填层析柱时应轻轻敲击管柱,使层析柱中的填充物装填结实,不能有缝隙,否则在层析的过程中会因淋洗液的作用形成断层或气泡,严重影响层析效果;在淋洗的过程中,应尽量使淋洗液顺着层析柱的管壁缓缓注入,避免破坏层析柱表面层的平整,使层析过程均匀连续,保证层析的效果。
2.3.2 标准混合溶液及样品定容溶剂的选择 在高效液相色谱(HPLC)分析中,样品的极性和进样的体积对色谱峰的峰型和保留时间有一定的影响。国家标准方法用正己烷定容标准混合溶液,但是在样品的处理过程中采用丙酮定容后进行液相色谱分析,由于溶剂的不统一,导致样品目标峰保留时间与标准溶液色谱峰保留时间有差异;在本方法的仪器条件下,进样量20μL,加标样品中苏丹红II和苏丹红III的保留时间比标准溶液保留时间推迟1min以上(图3A、B);在主要以保留时间定性的HPLC分析中,这种差异是致命的。考虑与流动相的互溶以保证结果的稳定性和重复性,本方法对苏丹红标准品和样品提取液均采用丙酮定容,所得目标峰的峰型尖锐对称,保留时间重现性好(图3C、D),定性准确。由图3还可以看出,实验采用基质固相分散—氧化铝层析法处理的样品非常干净,对目标峰无干扰,完全满足HPLC分析的要求。
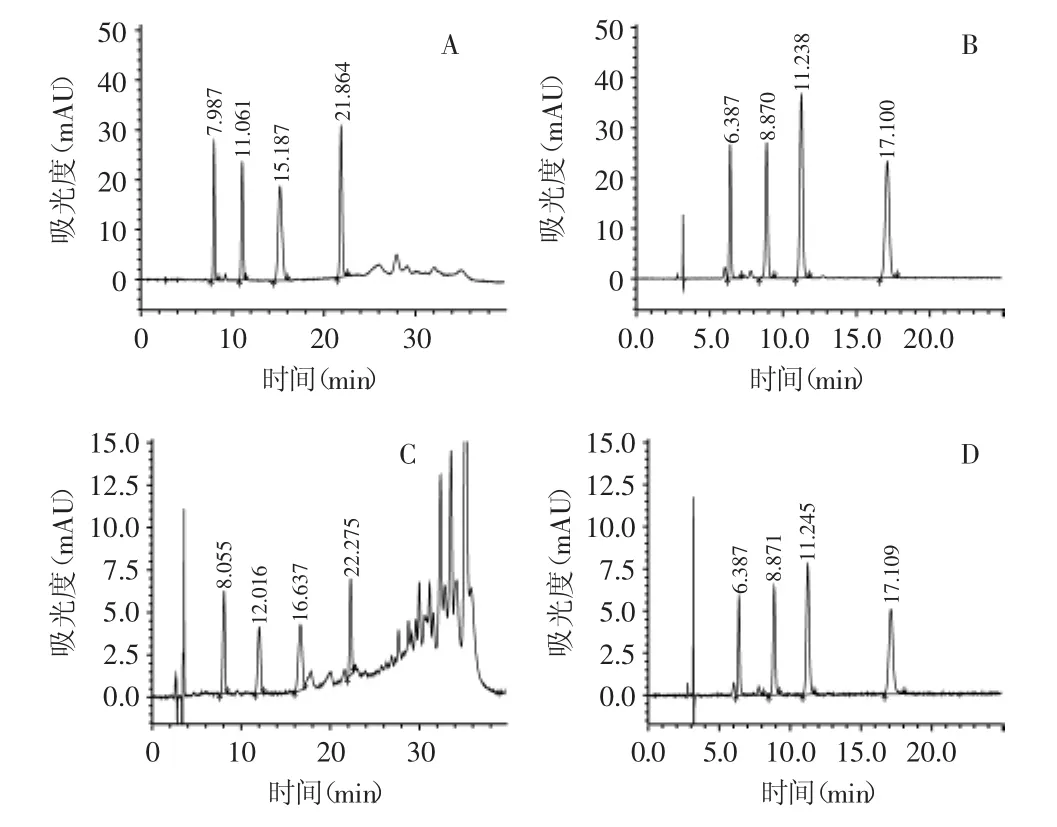
图3 两种方法标准溶液与加标样品色谱图Fig.3 Chromatogram of standard solution and spiked samples of the twomethods
2.3.3 氧化铝的活性及洗脱体积的确定 由于中性氧化铝对苏丹红的吸附活性是由含水量的多少决定的,一般先对中性氧化铝进行烘烤脱水活化,再加入一定量的水使其失活来控制中性氧化铝的活性。以含5%丙酮的正己烷50m L为洗脱液,测定不同含水条件下中性氧化铝净化加标样品的回收率。实验表明,当100g中性氧化铝中含水量低于1.5m L时,氧化铝活性太高,苏丹红I和苏丹红III不能完全洗脱,回收率较低;当100g中性氧化铝中含水量高于2.5m L时,氧化铝活性太低,对苏丹红的吸附力不够,用正己烷淋洗时部分苏丹红II和苏丹红IV就被洗脱下来,在收集的20m L淋洗液中测得苏丹红II和苏丹红IV约为加标量的40%~50%。经过多次调整和测定,最后确定在100g活化的中性氧化铝中加入1.8m L,所得的4种苏丹红回收率都较高。在此基础上,采用含5%丙酮的正己烷对加标样品进行洗脱,收集洗脱液,每5m L洗脱液收集一次,当收集的洗脱液总量在50m L以后,洗脱液中不含苏丹红,故洗脱液体积确定为50m L。
2.4 方法的线性范围、检出限和定量限
以质量浓度-峰面积做校正曲线,得到线性回归方程,4种苏丹红染料在0.10~50.00μg/m L范围内线性关系良好,相关系数均大于0.9999,线性方程见表1。以3倍信噪比为检出限(LOD),计算得出苏丹红Ⅰ、Ⅱ、Ⅲ、Ⅳ的检出限分别为0.008~0.011mg/kg。以10倍信噪比为定量限(LOQ),计算出苏丹红Ⅰ、Ⅱ、Ⅲ、Ⅳ定量限分别为0.025~0.033mg/kg。

表1 苏丹红Ⅰ~Ⅳ的标准曲线、检测限(LOD)和定量限(LOQ)Table 1 Standard curves,limitof detection,and limit of quantification for SudanⅠ~Ⅳ
2.5 方法的回收率和精密度
采用在空白样品中添加标准溶液的方法,对川味香肠中苏丹红染料进行3个浓度的加标回收实验,每个浓度重复6次。由表2可见,4种苏丹红染料在添加浓度为5.0~25.0mg/kg范围内平均回收率为85.54%~94.66%,相对标准偏差为0.87%~4.23%,完全满足食品分析的要求。其中,苏丹红Ⅰ的回收率最高,在5.0mg/kg的低浓度添加水平下平均回收率仍可达90.54%。苏丹红Ⅳ的平均回收率相对较低,相对标准偏差稍大,这是因为苏丹红Ⅳ的色谱峰峰型较宽,灵敏度偏低,特别是物质含量很低的情况下,在定量上不够精确。
2.6 样品测定
对市区的大中型超市和农贸市场的川味香肠进行抽检,分别按照上述方法进行处理,通过保留时间和光谱图定性,未发现苏丹红阳性样品。
3 结论
建立了川味香肠中苏丹红Ⅰ~Ⅳ的高效液相色谱分析方法。实验采用基质固相分散—氧化铝层析法对样品进行净化处理,高效液相色谱—二极管阵列检测器进行检测,利用光谱图和保留时间双重定性增加了方法的可靠性。与国家标准方法相比,减少了超声提取、过柱前的浓缩等操作步骤;由于简化了流动相并采用等度洗脱方式,基线平稳,所以定量更加准确,在缩短分析时间的同时减少有毒试剂的使用和排放。此方法简便、快速、可靠,重现性好,可用于川味香肠中苏丹红染料含量的测定。

表2 苏丹红在川味香肠中的回收率和相对标准偏差(RSD)(n=6)Table 2 Recoveries and RSDs of Sudan in Sichuan sausage(n=6)
[1]罗美中,李碧芳,何小青,等.高效液相色谱—二极管阵列法测定食品中苏丹红(I)着色剂[J].现代科学仪器,2005(5):54-56.
[2]Nohynek G J,Fantz R,Benech-kieffer F,et al.Toxicity and human health risk ofhair dyes[J].Food Chemistry and Toxicology,2004,42(4):517-543.
[3]Hahibi M H,Hassanzadch A,Mahdavi S.The effect of operational parameters on the photo catalytic degradation of three textile azo dyes in aqueous TiO2suspensions[J].Journal of Photochemistry and Photobiology A,2005,172(1):89-96.
[4]IARCWorking Group.IARC monographs on the evaluation of carcinogenic risk of chemicals to man:Some aromatic Azo compounds(Volume 8)[M].London:IARC,1975:125-129.
[5]励建荣,张蕾,丁献荣.HPLC快速测定辣椒及其制品中苏丹红Ⅰ号方法的建立[J].中国食品学报,2007,7(1):145-147.
[6]卫生部.《苏丹红危险性评估报告》[EB/OL].http://www.moh. gov.cn/,2005.
[7]侯晓林,孙英健,吴国娟,等.基质固相分散术-UPLC法检测禽蛋中对位红和苏丹红[J].食品科学,2010,31(24):285-288.
[8]吴银良,杨挺,赵健,等.固相萃取高效液相色谱法测定辣椒油中苏丹红和对位红染料[J].食品科学,2009,30(16):243-246.
[9]王明月,桂卫星,袁宏球.HPLC法测定咸蛋、皮蛋黄中的四种苏丹红染料[J].食品科学,2009,30(2):193-195.
[10]李军,雍炜,李刚,等.食品中苏丹色素的液相色谱分析方法[J].食品工业科技,2005,26(11):157-160.
[11]冯雷,孙文通,李军明,等.食品中苏丹红的HPLC/DAD/ MS分离分析方法研究[J].食品科学,2009,30(2):215-217.
[12]Mazzetti M,Fascioli R.Determination of 1-phenylazo-2-naphthol(SudanⅠ)in chillipowder and in chilli-containing food products by GPC clean-up and HPLC with LC/MS confirmation [J].Food Additives&Contaminants,2004,21(10):935-937.
[13]林佶,万玉萍,沈其萍,等.GC/MS定性定量分析食品中的苏丹红Ⅰ号[J].职业与健康,2006,22(22):45-46.
[14]李娜,董文宾,魏新军,等.紫外-可见分光光度法快速检测蛋黄中苏丹红IV的研究[J].食品工业科技,2009,30(12):397-400.
[15]蔡其洪,邹哲祥,李耀群.同步荧光法同时测定苏丹红Ⅱ和苏丹红Ⅲ[J].高等学校化学学报,2007,28(9):1663-1665.
[16]黄明元,彭晓君,李延志.共振光散射法检测辣椒及其制品中苏丹红Ⅰ[J].现代预防医学,2007,34(3):528-529.
[17]韩丹,于梦,吴梅,等.酶联免疫吸附分析法测定食品中的苏丹红Ⅰ号[J].分析化学,2007,35(8):1168-1170.
[18]GB/T 19861—2005食品中苏丹红染料的检测方法:高效液相色谱法[S].
[19]Barker SA.Matrix solid-phase dispersion[J].JChromatogr A,2000,885(1/2):115-127.
[20]Navarro M,Pico Y,Marin R.Application ofmatrix solidphase dispersion to the determination of a new generation of fungicides in fruits and vegetables[J].JChromatogr A,2002,968(1/2):201-209.
Determ ination of Sudan dyes in Sichuan sausage by combination of matrix solid-phase dispersion and high performance liquid chromatography
YANG Chun-lin1,LI Jia-jun1,HU Qiang1,WUW ei1,MENG Hui2,PENG Deng-shui3
(1.Center of Food and Chemical Product,Leshan Quality Supervising&Inspection Bureau,Leshan 614000,China;2.Shanghai Haifeng Modern Agricultural Co.,Ltd.,Shanghai202179,China;3.College of Resources and Environment,Sichuan Agricultural University,Ya’an 625014,China)
A high-performance liquid chromatog raphy(HPLC)method combined w ith matrix solid-phase d ispersion(MSPD)was estab lished for determ ination of Sudan I~IV in Sichuan sausage.The sam p le was m ixed with anhyd rous sod ium sulfate to be a part of the chromatography column.The samp les were c leaned up w ith hexane and rinsed w ith 50m L of acetone-hexane(5∶95,V/V).Separation was carried out on reversedphase InertsilODS-sp C18column(4.6mm×250mm,5μm)by using am ixture ofmethanoland form ic acid(999∶1)as mobile phase A,and 0.02mol/L ammonium acetate solution as mobile phase B(A∶B=85∶15,V/V)at a flow rate of 1.0m L/m in.Detec ted at the wavelength of 490nm by d iode array detector as the temperature was hold at 40℃.The retention time and spectra charac terization were both used to determ ine Sudan dyes and the content of Sudan dyes was quantified by external standard method.Sudan I~IV have good linearity in the range of 0.10~50.00μg/m L,the correlation coefficients were all above 0.9999 and the detection lim its of four Sudan dyes were 0.008~0.011mg/kg(S/N=3).With the add ition of 5.0 to 25.0mg/kg of Sudan standard to sam p les,the average recovery rates of four Sudan dyes in Sichuan sausage were between 85.54%and 94.66%.The relative standard deviations(RSDs)of sp iked sam p les varied from 0.87%to 4.23%(n=6).
Sichuan sausage;Sudan I~IV;high-performance liquid chromatog raphy;matrix solid-phase d ispersion
TS251.7
A
1002-0306(2012)09-0380-05
2011-08-11
杨春林(1982-),男,硕士,工程师,研究方向:食品质量与安全控制。
猜你喜欢
食品安全导刊(2020年14期)2020-12-04
红领巾·萌芽(2020年2期)2020-05-07
当代县域经济(2020年4期)2020-04-19
现代食品(2018年16期)2018-11-02
畅谈(2018年8期)2018-08-23
特别健康(2018年2期)2018-06-29
健康博览(2017年12期)2018-02-06
创新作文(小学版)(2016年16期)2016-11-11
食品工业科技(2016年17期)2016-10-31
创新作文(1-2年级)(2016年6期)2016-05-14
