
以分子动力组学的方法探究嗜盐酶的嗜盐机理
2013-03-03 05:26王四华黄可君张光亚
华侨大学学报(自然科学版) 2013年2期
王四华,黄可君,张光亚
(华侨大学 化工学院,福建 厦门361021)
嗜盐菌是一类生活在盐域环境中的微生物,主要生长在盐湖、盐沼、死海和盐场等环境中,Kushner等[1]将其分为弱嗜盐菌、中度嗜盐菌和极端嗜盐菌.嗜盐菌体内的蛋白酶不仅能耐受一定的盐离子强度,而且必需依靠一定的盐离子强度来维持其结构与功能的稳定 .对于极端嗜盐菌如嗜盐古生菌,其体内的酶可在盐浓度高达5.2 mol·L-1的环境下保持活性和稳定性[2],而在低盐浓度下,嗜盐古生菌体内的蛋白质会变性失活[3].研究表明:嗜盐菌主要依靠两条途径来维持胞内渗透压平衡[4-6],其一是细胞中聚集无机盐离子如K+,其二是聚集相容性物质如甜菜碱和四氢嘧啶等.尽管如此,有关嗜盐蛋白嗜盐机理尚未有确切的解释,而从原子层面系统了解蛋白分子在高盐条件下的动力学行为的研究也非常匮乏 .美国华盛顿大学Daggett教授的研究团队提出了动力组学(dynameomics)概念,并建立了动力组学数据库网站(http://www.dynameomics.org)[7],通过对一批具有代表性的蛋白质进行分子动力学模拟,以达到从原子尺度来理解蛋白质的功能和稳定性.分子动力学模拟越来越多地被应用于分子生物学方面,被用来理解、预测、模拟蛋白质、核酸的结构和性质,以及模拟DNA的双螺旋结构等[8].用分子动力学模拟的方法研究了嗜热蛋白与常温蛋白的分子动力学特性,可以从更微观的层面上探究嗜热与常温蛋白在结构柔韧性上的差异[9-11].然而,目前尚未见利用分子动力学模拟及组学的方法来探究嗜盐蛋白稳定性机理的报道 .本文通过对4组嗜盐蛋白及其同源非嗜盐蛋白的比较分子动力组学,模拟研究来阐明嗜盐酶的嗜盐机理.
1 材料与方法
1.1 数据获取
所利用的4组目标蛋白分别为二氢叶酸还原酶、苹果酸脱氢酶、碱性磷酸酶和核苷二磷酸激酶,每组分别包含一个嗜盐蛋白及其同源的非嗜盐蛋白.从蛋白质数据库(PDB)中获取目标蛋白,且这4组中的嗜盐蛋白在数据库(PDB)中的ID号分别为1VDR[12],2J5R[13],2X98[14]和2ZUA[15],对应的4个非嗜盐蛋白的ID号分别为7DFR[16],1UXI[17],1Y6V[18]和2VU5[19].因每组(嗜盐与非嗜盐)蛋白均为同一种酶,且它们蛋白质序列和空间结构非常相似,因此可以用来进行比较分子动力学模拟,以达到探究嗜盐酶稳定性机理的目的.
1.2 模拟方法
4组目标蛋白中的2J5R和2ZUA均含有4条链(A,B,C,D),1VDR,2X98,1Y6V及1UXI均含有两条链(A,B),7DFR及2VU5只含有一条链(A).分别只选取其中的一条链(A链)作为模拟对象,并将结构中多余的水分子去除;然后,将所有构建的体系分别置入同样大小的立方水体中,并中和每个体系的电荷,使得所有体系均为电中性.8条链所含氨基酸数目及体系最后所包含的原子数,如表1所示.
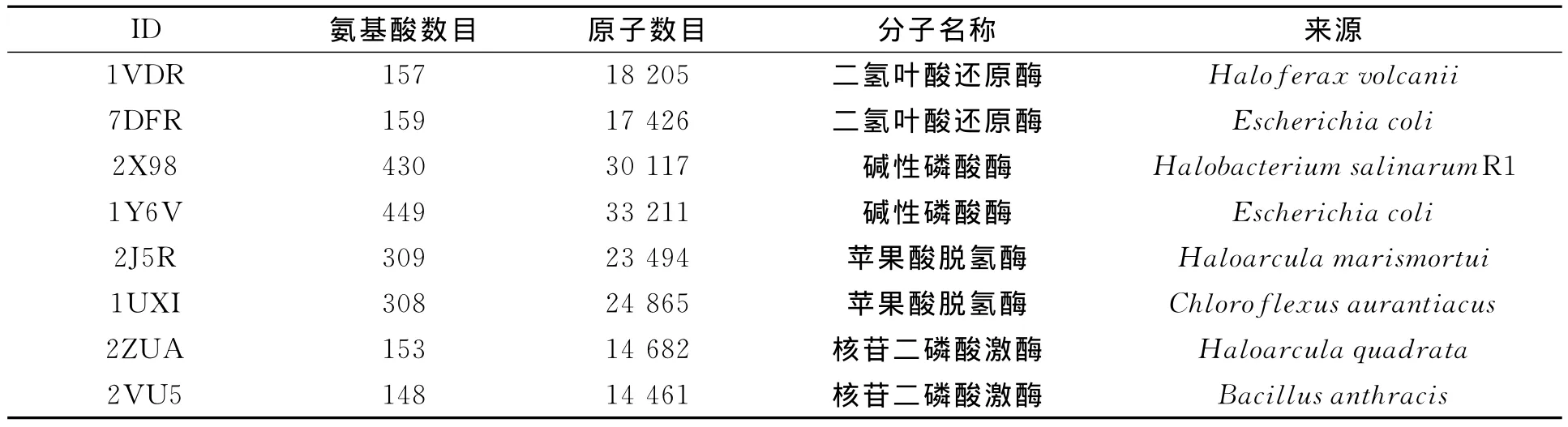
表1 8条A链所含氨基酸数目及体系最后所包含的原子数Tab.1 Amino acid number of eight A chains and the atomic number of the final system
将得到的8组体系分别进行10 ns的分子动力学模拟,一共进行了80 ns的分子动力学模拟.所使用的分子动力学模拟软件为NAMD[20]及与之相匹配的可视化软件VMD[21],选用CHARMm格式力场[22],所有模拟均在常压(101.325 k Pa)和常温(310 K)下进行,且分别采用Langevin Piston和 Langevin Thermostat方法控制压力和温度波动.采用PME[23]方法计算长程静电引力,非键相互作用力采用势能截断,截断半径为1.35 nm,对体系使用周期边界条件,使用SHAKE算法使水分子保持刚性.时间步长为2 fs,每1 ps输出一次计算结果,所有模拟均在等温等压系综下进行.
2 结果与分析
2.1 两类酶的分子动力学模拟所得参数分析
两类酶(嗜盐与非嗜盐)的溶剂可及性表面、盐桥、氢键(蛋白质分子内部及与水溶液形成的氢键)、末端距、回旋半径等参数,如表2所示.由表2可知:只有2X98所行成的盐桥比1Y6V的要少,这一反常差异可能是由于2X98与1Y6V氨基酸数目差异太大(二者相差19个氨基酸)造成的.除此之外,总体而言,嗜盐蛋白所形成的盐桥数目均比非嗜盐蛋白多,盐桥能消除盐离子的屏蔽效应,使分子结构具有刚性,对维持酶和蛋白质的三级结构的稳定起决定性作用.因此,可假定为嗜盐酶为了在高盐环境下维持其自身结构的稳定,所形成的盐桥数目比非嗜盐酶多.
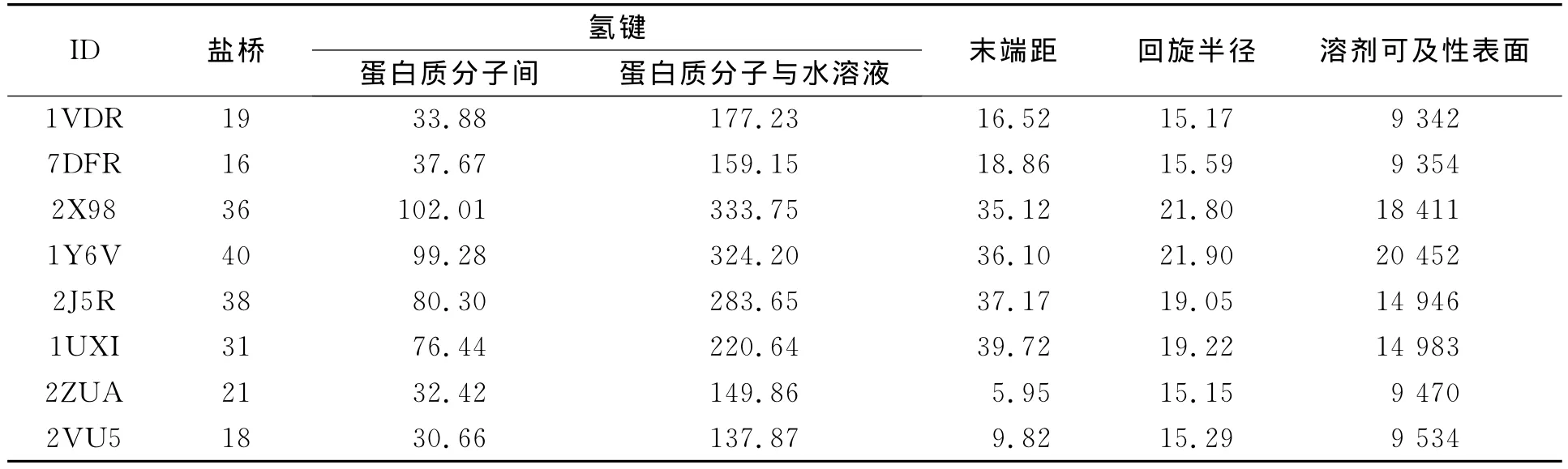
表2 嗜盐酶与非嗜盐酶之间各参数的比较Tab.2 Various parameters comparison of halophilic enzyme and non-halophilic enzyme
从数据上看,嗜盐酶另一突出特点,无论是其蛋白质分子内所形成的氢键还是蛋白质与外界水环境所形成的氢键,都比非嗜盐酶明显要多.有文献[24-25]表明,嗜盐酶所富含的酸性氨基酸残基可加强蛋白质与溶剂间的相互作用,为了适应高盐环境,嗜盐蛋白通过加强与溶剂间的水合能力来维持其结构与功能的稳定.对于嗜盐蛋白分子内所形成的氢键较非嗜盐蛋白多,这可假定为分子内的氢键在一定程度上可维持自身结构的刚性,因此也有利于嗜盐蛋白适应外界高盐环境.
文献[26]通过定点突变的方法发现,嗜盐蛋白通过降低其自身的溶剂可及性表面的机制来适应细胞外的高盐环境 .比较表2数据可以发现:嗜盐酶的溶剂可及性表面确实比非嗜盐酶的要小,在高盐缺水的环境下形成这一机制对嗜盐菌的生命活动是非常有利的.
末端距是用来表征蛋白质分子N端和C端的距离,回旋半径可用来表示蛋白质分子的紧密度.在这两项数值上,嗜盐酶同样比非嗜盐酶要小,这说明嗜盐酶在总体结构上较为紧密,嗜盐酶结构上的紧凑更有利于在高盐环境下维持其结构的稳定.
2.2 两类酶分子均方根偏差值的比较
均方根偏差(RMSD)是指模拟过程中两种蛋白质各部分原子偏离平均位置的程度,也就是原子运动幅度的大小,其值越大说明运动越剧烈,反映了蛋白质分子结构的刚性与柔性.图1为4组蛋白酶分子的RMSD值的比较.
从图1可知:4组酶中嗜盐酶的RMSD值均比非嗜盐的要小,嗜盐酶在结构上比非嗜盐酶更加刚性,这种特性也更加有利于其在高盐环境下保持结构的稳定.文献[10]通过分子动力学模拟的方法研究了嗜热酶与常温酶的稳定性机理,发现所研究的嗜热酶比常温酶的结构更具有刚性.对于是否大多数极端酶如嗜盐、嗜热、嗜压等结构均更具有刚性,则有待于进一步研究.
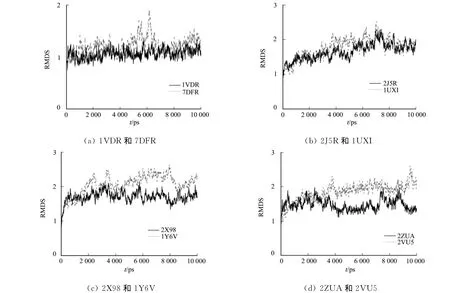
图1 蛋白酶分子的均方根偏差值比较Fig.1 Comparison of RMSD of the four group protein enzymes
2.3 两类酶各个氨基酸残基柔性比较
图2为4组蛋白质酶分子的每个氨基酸残基的RMSD值,可以直观地反应出具体部位的氨基酸残基数(n)波动幅度.从图2可知:非嗜盐酶氨基酸残基波动幅度比嗜盐酶要大,这与蛋白质整体的RMSD波动趋势相符.为了能更清楚区分嗜盐酶残基与非嗜盐酶残基的RMSD值的差异,将嗜盐酶残基的RMSD值减去非嗜盐酶残基的RMSD值,得到另一曲线,用diff-RMSD表示(图2).
比较1VDR与7DFR的每个残基波动情况,可发现7DFR在残基15~20上的RMSD值明显比1VDR要大,且二者在残基100~120间波动幅度均较小.很明显,1UXI在残基80~90上的RMSD值比2J5R要高出很多,而在残基65~70,95~110之间的RMSD值比2J5R却要小.比较2ZUA与2VU5的每个残基波动情况,可发现在残基40~60间2VU5对应残基波动幅度更大,同时发现二者C端的波动都很剧烈.1Y6V在N端及残基85~90,270~300,400~420区间上的RMSD值比2X98明显高出很多,而2X98在60~65残基上的RMSD值比1Y6V大.通过比较分析,发现非嗜盐酶多个部位的氨基酸残基柔性比嗜盐酶的要大很多,二者间的这些差异可能就是导致它们适应不同生长环境的一个因素.
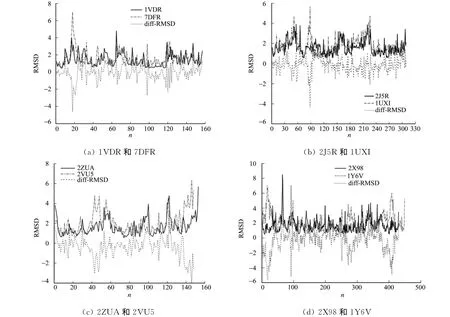
图2 每个氨基酸残基的柔性Fig.2 The flexibility of the amino acids
2.4 两类核苷二磷酸激酶三维结构的比较
为了进一步了解两类酶在三维结构上的差异,以核苷二磷酸激酶为例,对两类核苷二磷酸激酶进行高级结构的比对.通过在线结构比对工具(http:∥bioinfo3d.cs.tau.ac.il/MultiProt),对2ZUA 和2VU5的三维结构进行比对,发现二者除了在氨基酸40~60处有差异外,其他部位基本相叠合.比较图2中二者氨基酸残基柔性大小,可以发现在氨基酸40~60间二者出现了较大差异 .从图2中可看出:在氨基酸40~60处,2VU5的氨基酸残基柔性比2ZUA明显要大.
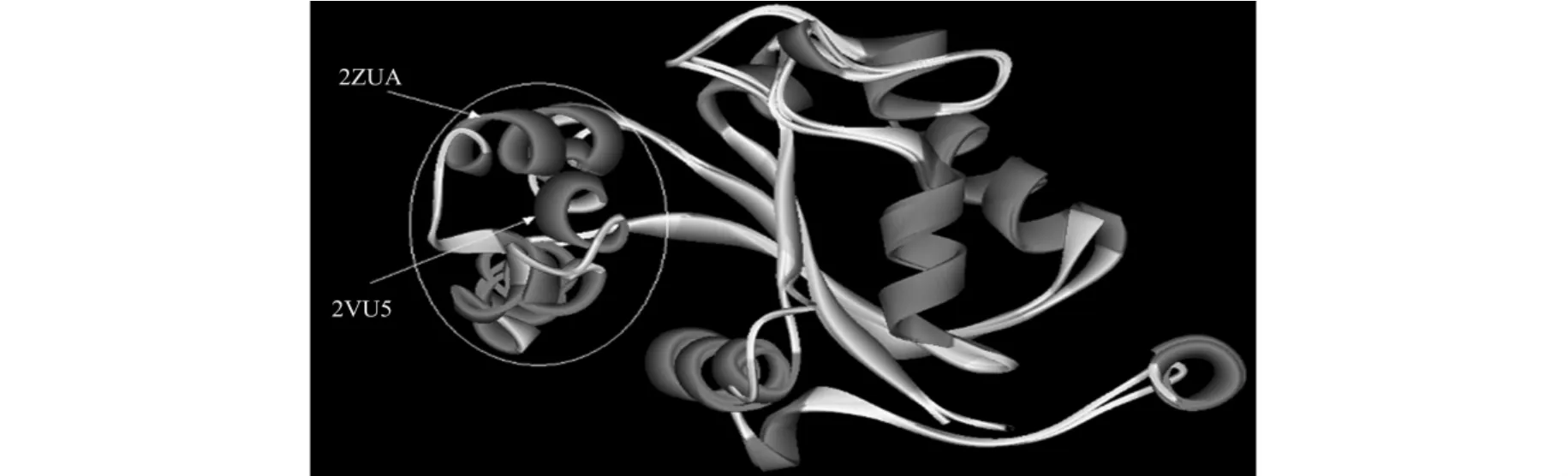
图3 2ZUA与2VU5三维结构的比较Fig.3 Comparison of three-dimensional structure of 2ZUA and 2VU5
巧合的是,通过比较二者的三维结构可发现它们正好在氨基酸40~60处出现明显的差异,如图3白色圆圈所示,二者在这一区域基本上为螺旋结构 .经仔细比较发现,2ZUA的螺旋结构比2VU5的更加紧凑,从而导致2ZUA的螺旋结构整体上比2VU5的要短.
二者这一区域的差异可能是导致它们适应不同生长环境的一个重要原因,2ZUA的螺旋结构更加紧凑可能会使得其结构更加稳定,从而能适应外界高盐环境.有趣的是,这一区域非常接近酶的活性中心,而是否与活性中心相互作用且二者的差异是否会导致引起二者活性中心的差异,以及二者的差异是否会导致它们整体结构上的差异,从而表现出不同的特性,有待于进一步研究.
3 讨论
有文献[27-28]报道嗜盐酶的酸性氨基酸含量比非嗜盐酶多,来自多个物种的嗜盐蛋白的平均等电点值为5.嗜盐蛋白分子表面带负电氨基酸残基较多[29-30],为了适应高浓度盐环境,嗜盐菌经过长期的进化导致其体内的蛋白质分子的疏水核心区较小[31].嗜盐蛋白表面的某些氨基酸与水合离子相互作用,有利于维持其构相的稳定[32-34].许多嗜盐蛋白的亚基表面存在一些特殊的盐离子结合位点[21],嗜盐酶与溶液中盐离子的结合一定程度上可以使其结构在高盐环境下保持稳定.对嗜盐硫解酶进行的研究表明:为了适应高盐环境,嗜盐硫解酶在一定范围内优先使用较小侧链的氨基酸,而在高盐溶液中,使用较小氨基酸残基能够明显降低分子的表面张力,有利于增加分子的灵活性[35].
理解嗜盐酶的嗜盐机理更有利于将嗜盐酶应用于工业生产中.Selim等[36]利用蛋白质组学技术来研究一株中度嗜盐菌的嗜盐机理,发现在不同梯度的盐离子强度下该菌机体内所产生的蛋白质种类有很大差异.嗜盐菌在环境生物治理、生物电子和医药工业等领域有着广泛的应用前景[37-39],其中的嗜盐酶也具有重大的应用价值.以色列Mevarechy研究小组将嗜盐的苹果酸脱氢酶和二氢叶酸酶基因在大肠杆菌中表达成功[40].文献[41]探索把嗜盐极酶用到提高从油井提取原油量的方法中,用嗜盐极酶可以分解掉瓜儿豆胶的粘性.
文中借鉴华盛顿大学Daggett教授的“动力组学”这一理念,分析了4组嗜盐蛋白及非嗜盐蛋白的动力学特性,从原子尺度来探究嗜盐酶的稳定性机理.对模拟得到的4组酶的运动轨迹进行分析,发现相比非嗜盐酶,嗜盐酶能形成更多的盐桥和氢键(无论是分子内氢键还是与溶剂所形成的氢键).盐桥及分子内的氢键在整体上能维持蛋白质结构的稳定,这对在高盐环境中嗜盐酶保持其结构的稳定是有利的.与溶剂形成较多的氢键,和文献[24-25]报道的嗜盐酶可加强与溶剂间的相互作用相符.
嗜盐酶的溶剂可及性表面较非嗜盐酶小,通过分析RMSD值、末端距及回旋半径等值发现嗜盐酶在结构上更具刚性,这一特性在应对外界高盐环境是非常有利的.通过比较分析发现,4组酶中非嗜盐酶很多部位的氨基酸残基柔性比嗜盐酶明显要大,猜测正是这一特性才导致嗜盐酶与非嗜盐酶适应不同的生长环境.
利用分子动力学模拟提供了一种新的研究嗜盐蛋白稳定性机理的方法,可为今后其他研究工作提供良好的借鉴.目前实验所得嗜盐蛋白晶体结构较少,仅收集到4组同源蛋白,虽然可以得到一些共性的规律,但随着蛋白质结构数据库的不断发展,相信今后可获得更多的样本,进行更大规模的蛋白质动力组学研究,使得所得的规律更具代表性,相关研究仍需不断深入.
[1] KUSHNER D J.Life in high salt and solute concentrations[M].London:Academic Press,1978:317-368.
[2] KAMEKURA M.Diversity of extremely halophilic bacteria[J].Extremophiles,1998,2(3):289-295.
[3] DOMINIQUE M,CHRISTINE E,GIUSEPPE Z.Halophilic adaptation of enzymes[J].Extremophiles,2000,4(2):91-98.
[4] VENTOSA A J,NIETO J J,OREN A.Biology of moderatelyhalophilic aerobic bacteria[J].Microbiolol Biol Rev,1998,62(2):504-544.
[5] GALINSKI E A,TRUPER H G.Microbial behaviour in salt-stresed ecosystems[J].FEMS Microbiol Rev,1994,15(2/3):95-108.
[6] AHARON O.Microbial life at high salt concentrations:Phylogenetic and metabolic diversity[J].Saline Systems,2008,4(2):(doi:10.1186/1746-1448-4-2).
[7] DAVID A C B,AMANDA LJ,SCHAEFFER R D,et al.Dynameomics:Mass annotation of protein dynamics and unfolding in water by high-throughput atomistic molecular dynamics simulations[J].Protein Eng Des Sel,2008,21(6):353-368.
[8] 欧阳芳平,徐慧,郭爱敏,等.分子模拟方法及其在分子生物学中的应用[J].生物信息学,2005(1):33-36.
[9] ERIC D M,WILLIAM W P,VALERIE D.Temperature dependence of the flexibility of thermophilic and mesophilic flavoenz ymes of the niteoedu-ctase fold[J].Protein Eng Des Sel,2010,23(5):327-336.
[10] TATYANA B M,ANNA V G,MARIA G K,et al.Flexibility and mobility in mesophilic and thermophilic homologous proteins from molecular dynamics and fold unfold method[J].J Bioinformatics and Computational Bio,2010,8(3):377-394.
[11] SANGEETA K,DEBJANI R.Comparative structural studies of psychrophilic and mesophilic protein homologues by molecular dynamics simulation[J].J Molecular Graphics and Modeling,2009,27(8):871-880.
[12] PIEPER U,KAPADIA G,MEVARECH M,et al.Structural features of halophilicity derived from the crystal structure of dihydrofolate reductase from the Dead Sea halophilic archaeon[J].Structure,1998,16(1):75-88.
[13] FIORAVANTI E,VELLIEUX F M,AMARA P,et al.Specific radiation damage to acidic residues and its relation to their chemical and structural environment[J].J Synchrotron Rad,2007,14(1):84-91.
[14] ANDY W,PATRIK J,RONNALD V,et al.Structural and biochemical characterization of a halophilic archaeal alkaline phosphatase[J].J Mol Bio,2010,400(1/2):52-62.
[15] ALOJORO Y,TAKEFUMI I,MASAHIRO K,et al.Molecular mechanism of distinct salt-dependent enzyme activity of two halophilic nucleoside diphosphatekinases[J].Biophysical Journal,2009,96(11):4692-4700.
[16] BYSTROFF C,OATLEY S J,KRAUT J.Crystal structures ofEscherichiacolidihydrofolate reductase:The NADP+holoenzyme and the folate-NADP+ternary complex:Substrate binding and a model for the transition state[J].Biochemistry,1990,29(13):3263-3277.
[17] ALEXANDRA B,BJORN D,DIMITRIOS M,et al.Large improvement in the thermal stability of a tetrameric malate dehydrogenase by single point mutations at the dimer-dimer interface[J].J Mol Biol,2004,341(5):1215-1226.
[18] WANG J,STIEGLITZ K A,KANTROWITZ E R.Metal specificity is correlated with two crucial active site residues inEscherichiacolialkaline phosph-atase[J].Biochemistry 2005,44(23):8378-8386.
[19] GAURI M,ANITA A,DIVYA D,et al.Crystal structure of theBacillusanthracisnucleoside diphospate kinase and its characterization reveals an enzyme adapted to perform under stress conditions[J].Proteins,2009,76(2):496-506.
[20] PHILLIPS J C,BRAUN R,WANG W.Scalable molecular dynamic with NAMD[J].Comput Chem,2005,26(16):1781-1802.
[21] HUMPHREY W,DALKE A,SCHULTEN K.VMD:Visual molecular dynamics[J].J Mol Graphics,1996,14(1):33-38.
[22] MACKARREL J,AD ashford,BELLOT D,et al.All-atom empirical potential for molecular modeling and dynamics studies of proteins[J].J Phys Chem,1998,102(18):3586-3616.
[23] DARDEN T,YORK D,PEDERSEN L.Particle mesh ewald:AnN~log(N)method for ewald sums in large systems[J].J Chem Phys,1993,98(12):10089-10092.
[24] COSTENARO L,ZACCAI G,EBEL C.Link between protein-solvent and weak protein-protein interactions gives insight into halophilic adaptation[J].Biochemistry,2002,41(44):13245-13252.
[25] TARDIEU A,BONNETÉF,FINET S,et al.Understanding salt or PEG induced attractive interactions to crystallize biological macromolecules[J].Acta Crystallogr Sect D Biol Crystallogr,2002,58(10):1549-1553.
[26] TADEO X,LÓPEZ-MÉNDEZ B,TRIGUEROS T.Structural basis for the aminoacid composition of proteins from halophilic archea[J].PLoS Biol,2009,7(12):e1000257.
[27] BENACHENHOU N,BALDACCI G.The genes for a halophilic glutamate dehydrogenase,sequene transcription and phylogenetic implications[J].Mol Gen Genet,1991,230(3):345-352.
[28] BALDACCI G,GUINET F,TILLIT J,et al.Functional implications related to the gene structure of the elongation factor EF-Tu fromHalobacteriummarismortui[J].Nucl Acids Res,1990,18(3):507-511.
[29] MEVARECH M F,FROLOW L M.Halophilic enzymes:Proteins with a grain of salt[J].Biophys Chem,2000,86(2/3):155-164.
[30] EISENBERG H.Life in unusual environments:Progress in understanding the structure and function of enzymes from extreme halophilic bacteria[J].Arch Biochem Biophys,1995,318(1):1-5.
[31] COQUELLE N,TALON R,JUERS D H.Gradual adaptive changes of a protein facing high salt concentrations[J].J Mol Biol,2010,404(3):493-505.
[32] FROLOW F,HAREL M,SUSSMAN J L.Insights into protein adaptation to a saturated salt environment from the crystal structure of a halophilic 2Fe-2S ferredoxin[J].Nat Struct Biol,1996,3(5):452-458.
[33] ZACCAI G,CENDRIN F,HAIK Y,et al.Stabilization of halophilic malate dehydrogenase[J].J Mol Biol,1989,208(3):491-500.
[34] EBEL C,COSTENARO L,PASCU M,et al.Solvent interactions of halophilic malate dehydrogenase[J].Biochemistry,2002,41(44):13234-13244.
[35] 刘铁汉,周培瑾.极端嗜盐硫解酶基因的克隆和氨基酸组成分析[J].微生物学报,2002,42(4):406-410.
[36] SELIM C,BERNA S A,AZIZ A D,et al.Proteomic insight into phenolic adaptation of a moderately halophilicHalomonasp.strain AAD12[J].Can J Microbiol,2011,57(4):295-302.
[37] LIANG Chen-hui,YI Wei,LI Bin.Case study of hypersaline organic wastewater treatment with SBBR process[J].Pollution Control Technology,1998,14(4):226-228.
[38] BIRGE R R.Photophysics and molecular electronic applications of the rhodopsins[J].Annu Rev Phys Chem,1990,41:683-733.
[39] SHI Hai-ping,SU Tao.Study on mcrobiol frmentation of cllecting for ply-β-hydroxybutyrate[J].Food and Fermentation Industries,1998,24(2):79-82.
[40] EICHLER J.Biotechnological uses of archaeal extremozymes[J].Biotechnology Advances,2001,19(4):261-278.
[41] WANG Shi-fen,YANG Jian.Treatment of petro-fermentation wastewater with high salt content[J].Water and Wastewater Engineering,1999,25(3):35-38.
猜你喜欢
生物化学与生物物理进展(2022年6期)2022-07-21
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19
中国洗涤用品工业(2019年4期)2019-05-11
中成药(2018年1期)2018-02-02
中学化学(2015年12期)2016-01-19
池州学院学报(2015年3期)2016-01-05
动物医学进展(2015年10期)2015-12-07
天津科技大学学报(2015年2期)2015-08-09
食品工业科技(2014年7期)2014-03-11
原子与分子物理学报(2014年3期)2014-02-28
