
基元模式分析在生物网络和途径分析中的应用
2013-06-30 08:25赵权宇于水燕史吉平
生物工程学报 2013年6期
赵权宇,于水燕,史吉平
中国科学院上海高等研究院生物炼制实验室,上海 201210
与生物代谢网络相关的系统生物学计算方法包括有约束代谢通量分析(Constraint-based metabolic flux analysis)和代谢途径分析(Metabolic pathway analysis)等[1-3]。代谢途径分析研究代谢网络中从底物到产物的可能途径或内部代谢产物的循环。代谢途径分析的多数算法都是以凸分析为基础的,主要包括基元模式(Elementary mode)和极端途径(Extreme pathway)[4]。基元模式分析允许代谢网络模型中同时存在可逆和不可逆反应或传递过程;极端途径分析需要把代谢网络内部的可逆反应拆分成两个不可逆反应。关于基元模式和极端途径的关系曾经有过争论,目前较为一致的看法是极端途径为基元模式的子集[5]。基元模式分析是应用最多的代谢途径分析方法。文中综述了基元模式分析的最新进展,并探讨了未来的重要发展方向。
1 基元模式分析的算法与软件
基元模式是代谢网络中所有可能代谢途径的集合,基元模式的数目随着代谢网络规模的扩大急剧增加,将会引发组合爆炸(Combinatorial explosion)。对中等规模代谢网络,基元模式数就可能超过200万,而基因组尺度代谢网络的基元模式很容易就超过2600万[6]。这给基元模式的计算和分析都带来了困难。Terzer 等[7]提出位模式树算法加快基元模式的计算速度。其显著特征是在正常变量中区分极端线,引入随机排序方法加强并行计算,提出余数方法改善求秩,在硬件上使用多核技术。该算法用Java 开发,可以在Matlab 中运行,成功计算了大肠杆菌中心碳代谢网络(106个反应)的2638万个基元模式,以及基因组尺度幽门螺杆菌(381个反应)的500万个基元模式。Jevremovic 等用C++开发的算法先对代谢网络进行压缩,再用并行计算的方法计算酿酒酵母(80个反应)的1332万个基元模式,可以在0.5 h 左右完成[8]。这些算法还有待进一步改进,比如继续优化数据结构和改善求秩等。已经证明,基元模式的计算是NP-Complete 问题(多项式复杂程度的完全非确定性问题,Non-deterministic polynomial-complete problem)[9-10]。虽然提高计算速度可以缩短计算时间,对多数研究者来说,分析1000万个基元模式仍旧是困难的。KEGG (Kyoto Encyclopedia of Genes and Genomes,KEGG)数据库根据功能不同将代谢网络分解成若干的子单元并提供代谢途径图,如糖酵解和三羧酸循环等。这些子单元包括的代谢反应或过程一般不会超过300个,计算子单元的基元模式是相对容易的。Schwartz等[11]最早尝试从这些子单元分析基因组尺度酿酒酵母代谢网络模型的基元模式,并结合基因表达数据分析了不同压力条件下的代谢功能。基元模式最多的子单元是戊糖磷酸化途径,也仅有206个基元模式,给基元模式分析带来很大方便。该方法虽然无法将子单元与全网络尺度的基元模式建立有效的联系,但已经初步获得了基因组尺度的基元模式与转录的关系。Kaleta 等同样希望通过分析子系统来获得基因组尺度代谢网络的潜在代谢途径[12]。他们的出发点是在子网络分析中如果外部代谢物选择不当,可能丢失一些关键的基元模式。因而提出基元通量趋势(Elementary flux patterns)的概念,这些基元通量趋势是一个子系统中反应的子集,代表大网络每个稳态通量分布在子系统上的可能途径。目前可以考察最小培养基,计算最小剪切组和考察鲁棒性等,进一步的研究还在进行中。另外一种策略是开发新的算法,避免计算全部的基元模式。de Figueiredo 等[13]提出的是K 次最短基元模式的算法,这里的K 不是路径长度,而是代谢网络中基元模式的路径长度排序。比如,三羧酸循环的1次最短基元模式(K=1)的路径长度是2。应用K次最短基元模式算法可以挑选基因组尺度大肠杆菌和谷氨酸棒杆菌代谢网络模型中与赖氨酸生产相关的基元模式的子集,如何建立K 次最短基元模式与代谢行为的关系还有待深入研究。Song 等[14]用产率分析算法计算基元模式的子集。将包含38个反应的重组酿酒酵母的基元模式数从369个降低到35个。他们的新算法基于凸包分析,难以用于多维体系,仅对小规模的代谢网络适用。Rezola 等[15]使用了类似的策略,他们提出了产生通量模式(Generating flux mode)的概念,计算构成通量空间的边。对基因组尺度(2082个反应,1668个代谢物)的大肠杆菌代谢网络,该算法可以计算K 等于100的赖氨酸产生途径的产生通量模式。该算法基于优化方法求取凸基,缺陷是产生通量模式并不唯一。在随机选定产生通量模式时,可能忽略关键的基元模式,影响对代谢网络结构的解析。即使基元模式只有几千个,手动分析这些基元模式仍是困难的。借助普通模体集聚(ACoM)可对基元模式进行聚类分析[16],不过也仅适用于小型网络。依据具体基因型和环境条件,约束代谢网络空间,有助于减少基元模式数目。如Ferreira 等开发了集成环境组学数据的隐途径投影算法(Projection to latent pathways),整合环境组数据求取BHK 细胞的有效基元模式[17]。这仍需要先计算所有的基元模式,再进行简化。应用环境实验数据在基元模式上进行多变量回归时,原则上实验变量数要高于显著的基元模式数,可对于大规模网络来说很难实现。
软件在系统生物学的研究中有着重要的作用,性能良好的软件需要具有功能丰富、界面友好、计算速度快和方便调用等特点。目前,CellNetAnalyzer[18]、OptFlux[19]、YANAsquare[20]、EFMtool[7]和Tools-4-Metatool (T4M)[21]等都可以计算基元模式。CellNetAnalyzer 是基于Matlab的图形界面软件,可以计算基元模式以及分析相关的代谢网络拓扑性质,同时还可以研究信号传导网络。为解析高通量数据,CellNetAnalyzer 也开发了可以独立于图形用户界面的应用程序界面[22]。YANAsquare为Java开发的图形界面软件,调用Metatool 作为计算基元模式的工具,提出的基元模式活性与酶活的转换关系缺乏理论基础,在应用中需要注意。YANAsquare 的优点是可以实现代谢网络模型SBML 格式和Metatool 格式的转换,但是计算速度慢,数据格式还有待改进。比如,目前化学计量系数只能为整数,而很多代谢网络的生物质合成反应系数并非整数。T4M 是基于网络的基元模式分析平台,既可进行基元模式分析,也可以进行比较,其代谢网络及基元模式的图形自动输出还有待优化。还需要注意的是,T4M 的输入和输出是Metatool 格式,并非SBML 格式,需要先用YANAsquare 等软件将SMBL 格式转化为Metatool 文件。EFMtool 在Matlab 中运行,可以计算基因组尺度代谢网络模型,但不是图形界面文件,略显不便。OptFlux也是用Java 开发的软件,可以计算基元模式,然而其他的代谢途径分析功能不多。COBRA 中也有了计算基元模式的模块,但是其主要功能还是进行代谢网络构建与代谢通量分析[23]。
2 基元模式分析的应用
基元模式分析在代谢工程与生物过程工程等领域的应用可以归纳为以下6个方面(图1)。
2.1 代谢途径与鲁棒性

图1 基元模式分析的应用Fig.1 Applications of elementary mode analysis.
构建代谢网络,计算基元模式仅是基元模式分析的第一步。还要通过这些基元模式分析代谢途径的功能。Klamt 等建立了紫色非硫菌红螺菌电子传递链的动态网络模型[24]。该模型在稳态下包含9条基元模式,分别对应光合成和呼吸过程中的ATP 合成与可逆电子流动以及延胡索酸还原。一般情况下,三羧酸循环、糖酵解和糖异生途径中没有将乙酰辅酶A 转化为葡萄糖的基元模式[25]。如果插入异柠檬酸裂合酶和苹果酸合酶相关基因,就可以实现脂肪到葡萄糖的合成。基元模式可以分析基因和环境扰动对微生物的影响。通过基元模式分析发现,厌氧条件下戊糖磷酸化途径对克雷伯氏肺炎杆菌以甘油为底物生成1,3-丙二醇有重要影响,而在好氧条件下三羧酸循环的影响更大;降低氧的消耗速率有利于1,3-丙二醇的合成[26]。Taffs 等将基元模式的概念用于微生物群落中物质和能量流动的分析[27]。具体分析了美国黄石公园热泉中光合细菌、不产氧光合细菌(绿曲挠菌等)和硫还原细菌间在白天和夜晚的相互作用,包括生物质合成和能量产生以及固氮效率等。通量平衡分析(Flux balance analysis,FBA)是在选择特定目标函数后通过线性规划求取通量平衡分布。如果目标函数是目标代谢物最大产率,FBA 预测得到的目标代谢产物产率往往都偏高,而通过基元模式分析可以解释植物乳杆菌的实验室适应性进化[28]。除了应用到细菌和酵母的代谢途径分析,基元模式分析在微藻生物燃料的分析中也可以发挥作用[29-30]。小球藻先进行自养固定CO2,再进行异养产油。其中包括Rubisco 途径的基元模式得到强化,可以解释为何产油效率的提高[29]。Rugen 等根据文献上的目标函数对代谢通量进行分解,分析了莱茵衣藻光合自养条件下的代谢,结果表明光输入在代谢响应中有重要作用[30]。
代谢网络具有鲁棒性(Robustness)或健壮性(Redundancy)。大量的实验数据证明,除个别关键基因的敲除会引发严重后果,多数的单基因敲除操作仅会明显改变代谢网络的局部代谢通量分布,而对网络整体的影响不大。生命体可以通过调控来降低基因敲除的影响,维持自身的稳定。这也是生命体在长期进化发展中逐步形成的。代谢网络结构的复杂性是其鲁棒性的基础之一。代谢网络的鲁棒性可以由网络结构及其动力学、基于优化的方法和代谢途径分析等计算[31]。在代谢网络中,由某底物到某产物的基元模式通常有很多,这就保证其特定的功能不会被轻易地破坏。在特定生理条件下,只有少量基元模式具有显著活性。多数基元模式看似冗余,却有可能在基因和环境扰动情况下被激活,是生物体维护自身稳定的需要。基元模式数目可以作为标度网络鲁棒性的一个指标,随着基因[32]或路径[33]敲除数目的增加,鲁棒性自然降低。最小切割子集也可以表示网络的冗余性。Haus 等最早开发的算法是先计算基元模式,再计算最小切割子集,计算步骤多[34]。在新算法中由代谢网络结构信息直接计算最小切割子集,耗时大大减少[35]。生物或催化系统的冗余性和稳定性联系紧密。在一个简单的自维持系统中,三个基元模式有不同的意义,删除一个都无法维持系统的稳定[36]。
2.2 代谢通量分解
代谢通量可以分解在基元模式上,并表示为基元模式的线性集合。代谢通量分解是代谢网络分析的一种重要手段,可以由实测的代谢通量分布求取基元模式系数,从而判断该实验条件下的主要基元模式,并分析代谢网络的复杂行为。根据热力学原理,代谢网络内部的循环对代谢通量没有贡献。代谢通量是基元模式的非负线性组合。

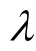
如果矩阵Pexp的秩接近基元模式数,不同目标函数的计算结果差别不大;反之,就会得到差别显著的解,这就需要目标函数有好的生物学或者物理学意义,才能具有良好的适用性。线性规划求取基元模式系数与FBA 相似,最大生物量作为目标函数未必适合所有生物及其生理条件。最小范数和最小基元模式数并没有明确的生物意义。最大基元模式数假设在外界扰动后代谢网络趋向冗余,但是在非零基元模式数相同的条件下,没有确定基元模式系数如何分配。最大信息熵原理广泛应用于物理、化学和生态领域,以及生物信息学中的基因表达与序列分析。应用最大生物量增长、最小范数和最大信息熵等目标函数一起比较分析中国仓鼠卵巢癌细胞、大肠杆菌和酿酒酵母的代谢通量分布,最大信息熵原理算法得到最可行的基元模式系数分布,在酶控通量算法中也可以得到更准确的代谢通量分布[37]。基元模式系数的优化算法[38-40]见表1。除选择优化中合适的目标函数,添加热力学信息作为优化的附加限制条件,也可以消除不可行的基元模式[40-41]。
熵是系统非均匀性的标度,和系统的变化趋势相关。Walschin 等率先分析了代谢途径热力学,并关联了基元模式系数及其燃烧熵[42]。该文的缺陷之一是无法计算所有胞内代谢通量。另外,为得到简单线性关系,该文用基元模式家族(Family of elementary mode)的概念简化了代谢网络,忽略了生物质的合成,用化合物理想状态燃烧熵计算基元途径集总反应的熵,未进行生物体系溶液非理想性修正,结论为大肠杆菌厌氧过程趋向熵最小。之后,该文作者重新进行了分析,认为代谢网络及其进化趋向最大化学熵[43]。最近,他们又对适应进化后的最大熵变化进行了讨论[44-45]。
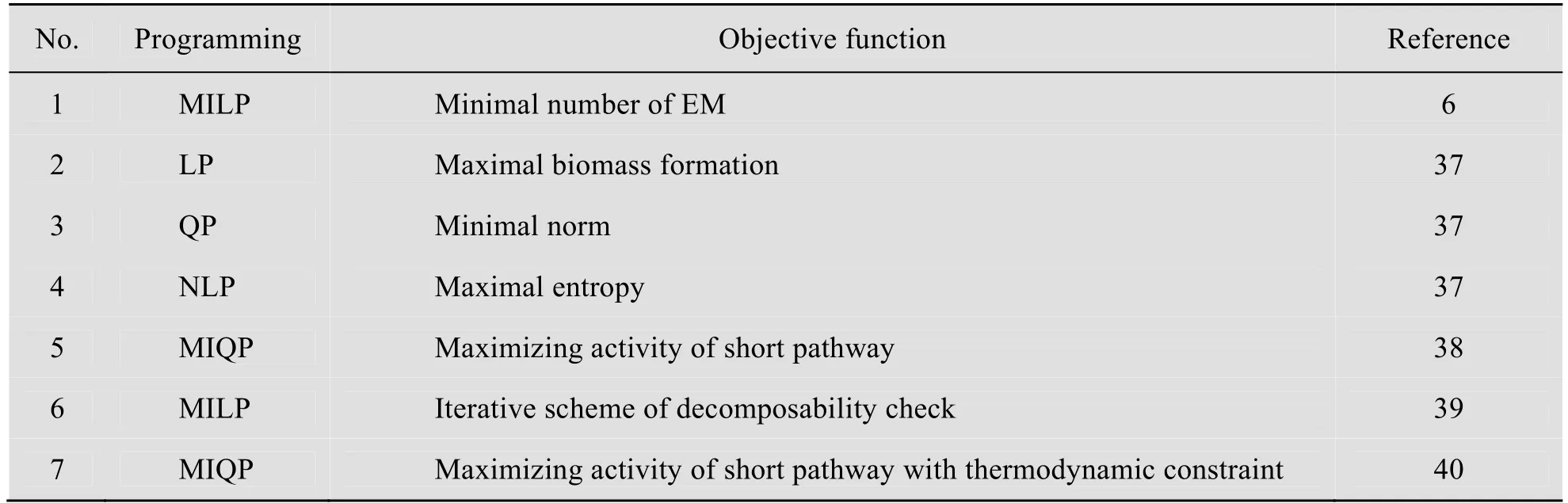
表1 基元模式系数优化算法Table 1 Optimization algorithms of elementary mode coefficients
本文作者引入最大信息熵(Shannon entropy)优化基元模式系数[37],与酶控通量算法结合,成功预测大肠杆菌基因和环境扰动后的代谢通量分布。在欠定和不确定条件下,通过胞外13C 同位素标记代谢通量数据估算杂交瘤细胞、枯草芽胞杆菌和大肠杆菌胞内代谢通量分布。与其他目标函数相比,可以更为可靠和可行的估算胞内代谢通量分布[46]。再应用拉格朗日算子来最大化基于熵的目标函数,分析求解基元模式系数[47]。用最大熵原理优化基元模式系数阐明了基元模式与代谢通量的关系,基元模式反映了代谢网络的微观性质,代谢通量反映了代谢网络的宏观性质,宏观性质是微观性质的加合,而且这种加合服从最大信息熵原理。最大熵原理作为目标函数优化基元模式系数为基于基元模式的代谢通量预测提供了可靠基础。信息熵与化学熵的关系有待深入探索。
图2是用线性规划和最大熵原理求得的酿酒酵母代谢网络的基元模式系数[37]。图中颜色表示对应基元模式系数的绝对偏差。在原文献中以柱状图表示取得的基元模式系数,而图2可以反映这些基元模式系数的差异。可以看出两个目标函数求取的多数基元模式系数值很低,因此偏差也小,表现为白色。最大熵原理求得的第31条基元模式的系数最大,而二次规划求得的第28条基元模式的系数最大,但是这两个基元模式系数差别较大,在图中表现为深色条带。
2.3 稳态代谢通量分析

图2 最大熵原理(MEP)和二次规划(QP)求取基元模式系数(酿酒酵母,μ=0.3)Fig.2 Elementary mode coefficients calculated by Maximum Entropy Principle (MEP) and Quadratic Programming(QP) for S.cerevisiae (μ=0.3).
在系统生物学中,有约束代谢通量分析用以考察代谢产物分布以及代谢网络中的关键节点等。代谢通量可以通过动态或稳态模型计算。动态模型需要大量实验数据关联动态模型参数,一般来说包括的生化反应数少。代谢控制分析被用于关联酶活和代谢通量的关系。代谢控制分析的缺点是需要大量、准确的实验数据或者代谢网络内部所有生化反应的动力学方程。因此,目前局限在简单代谢途径或小尺度的代谢网络分析中。
有约束代谢通量分析是常用的代谢网络稳态分析方法,特别是FBA。在基因组尺度的代谢网络分析中,FBA 成功地用于大肠杆菌、酿酒酵母、枯草芽胞杆菌等微生物,中国仓鼠卵巢癌细胞(CHO)和幼鼠肾细胞(BHK)等动物细胞和小球藻等植物细胞的代谢网络分析。FBA 计算简单,应用广泛,问题是必须事先指定目标函数,再通过线性规划求取代谢通量分布。最大生物量增长可以作为目标函数计算大肠杆菌等微生物的代谢通量分布,并能与实验数据基本符合。但是这个目标函数不能适用于全部的生命体,以及一些生物体的不同生长阶段。比如,人的红细胞本身就不进行生物质合成,无法选择最大生物量作为目标函数。微藻在正常培养和氮缺乏条件下的生物质中糖、蛋白质和脂类等主要组成发生显著变化,特别是氮胁迫后生物质合成受到抑制。以最大生物量增长为目标函数显然不适合确定微藻不同调控状态下其生物质内糖、蛋白质和脂类的具体分配。另外,最大ATP 利用等基于生物学假设的目标函数的通用性还要考察。
基于基元模式分析的代谢通量算法研究是目前代谢工程领域的新增长点。酶控通量算法(Enzyme control flux)[48]集成酶活数据和野生株代谢通量数据预测突变体的代谢通量分布。与FBA 不同,不需事先指定目标函数,可以集成实验数据对代谢网络进行分析。基因改变通量算法(Genetic modification of flux)[49]联系代谢网络结构、功能和调控,对酶活改变后的代谢通量分布进行预测,与实验报道符合良好。基因改变通量算法的核心是由野生株和突变体的相对基因表达和野生株基元模式系数预测突变体的基元模式系数,进而预测突变体的代谢通量分布。该算法可在基元模式水平上定量理解由于基因或环境扰动造成的代谢或生理状态的变化[47]。同时也说明了酶控通量算法是集成酶活数据预测代谢通量分布的可行框架。
2.4 动态模型与生物过程模拟
疫苗和基因重组药物主要来自于微生物和动物细胞的生物反应器培养。生物过程的动态模拟是过程分析、检测和控制的重要工具。生物过程是复杂的,除了受外界环境因素的影响,生命体本身也具有自我调节作用。经验模型往往不能够反映生物过程的内部规律。目前的检测技术测定的代谢物有限,在线测定胞内代谢物的变化也有困难。随着基因组技术的完善,对生物代谢过程的了解不断深入。基元模式也是生物过程建模的手段之一。
除了内部循环,每个基元模式都有一个集总反应来表示该条基元模式从外源底物到外源产物的计量关系。这些集总反应从基因水平描述了生物过程的可能途径,为建立代谢网络动态模型和生物过程模型提供了基础[50]。建立生物过程模型需要有准确的计量学代谢网络模型,通过基元模式分析确定外部代谢物构成的集总反应,再经过简化建立宏观反应模型,最后由实验数据拟合动力学参数,得到动力学模型。目前,基于基元模式的生物过程动态模型已成功应用于CHO 细胞[51]、杂交瘤细胞[52]和BHK 细胞[53]等的反应器培养过程,用以生产单克隆抗体或重组融合糖蛋白。目前这些基于基元模式的生物过程模型都是单细胞模型,也未考虑细胞周期的影响。
普度大学Ramkrishna 教授建立了基元模式和控制论变量的混合模型[54-59]。这类模型的特点是通过控制论描述外部代谢物的动态变化,而由基元模式估算稳态下的胞内代谢通量分布。他们首先用包含23个反应的代谢网络模型分析了大肠杆菌GJT001的厌氧代谢,与通量平衡分析比较可以更好地预测琥珀酸的生产[54]。并预测了大肠杆菌pta-ackA 基因敲除突变株YBS121的厌氧发酵过程,以及基因扰动对3株表达外源基因的大肠杆菌突变体的影响(表达博伊丁假丝酵母Candida boidinii 的fdh1基因、pta-ackA 敲除同时外源插入枯草芽胞杆菌 alsS 基因,以及adhE-idhA 敲除同时外源插入乳酸乳球菌pyc 基因),可以与实验结果较好吻合,问题是所用的大肠杆菌模型仅有12个反应[55]。由于采用的大肠杆菌模型过于简单,他们又分析了酿酒酵母在葡萄糖和木糖双底物条件下的发酵[56],与宏观反应模型(MBMs)和动态通量平衡分析(dFBA)相比,混合控制论模型(HCM)可更清晰地阐述代谢控制机制。酿酒酵母的代谢网络包含38个反应,反应数增加一般来说基元模式数也会增加,会造成过参数化的问题。他们用产率分析算法[14]对基元模式做简化,降低需要拟合的参数。之后他们又开发了集总混合控制论模型(L-HCM),将经典的集总控制论模型和HCM 结合在一起,从有限的数据预测酿酒酵母(代谢网络模型包含53个反应)的好氧发酵[57]。进一步修改集总权重的计算方式并将L-HCM 推广到大肠杆菌(代谢网络模型包含67个反应)的厌氧生长[58]。最近,该模型又研究了真氧产碱杆菌(代谢网络模型包含36个反应)合成聚羟基丁脂的情况,认为多稳态在实际中发生的可能性很小[59]。这类基于基元模式的混合模型在预测代谢行为上有一定的优越性,但是和dFBA 相比还无法用到基因组尺度的代谢网络模型上,主要原因还是基元模式的组合爆炸问题。
2.5 代谢调控
生物网络结构、功能、调控间存在紧密的联系。控制有效通量(Control effective flux)是基于基元模式的算法,仅仅需要代谢网络结构信息就可以关联环境条件扰动(如底物由葡萄糖换成乙酸、乳酸或乙醇)后转录水平的变化[60]。本文作者将控制有效通量算法用于基因扰动,比如大肠杆菌的基因敲除,同样可以得到较满意的关联结果[49]。为提高关联精度还可以改进算法,如使用更大规模的代谢网络、根据不同的培养条件选择主要的基元模式等[60]。控制有效通量算法的局限与代谢网络结构有关,比如仍无法区分同工酶。Schuster 等首先否定了代谢网络向分子产率最大的方向进化[61]。Jens Nielsen 和合作者提出应用基于化学熵的算法选择酿酒酵母代谢过程中控制通量的关键酶[62]。最大化学熵和最大信息熵有相似的形式。信息熵在没有足够信息的情况下适用,而化学熵要对含生物体系溶液进行非理想性修正,否则可能会影响预测结果的准确性。代谢途径的熵分析已经引起很多专家的重视,值得深入研究。
环境因子对生物的影响也是显著的。大量研究专注于基因功能和基因-基因相互作用,而基因和环境的相互作用研究却不多。这些影响可以从多方面进行考虑。比如,细胞培养过程中存在不同生理状态,而这样的状态与培养基中细胞分泌的代谢物浓度等存在一定联系。环境组引导的潜在途径投影算法(Envirome-guided projection to latent pathways)用基元模式描述细胞功能,关联BHK 细胞在不同培养条件的27个动态环境数据(温度、pH,渗透压和24个胞外代谢物浓度)可以获得在统计学上最显著的基元模式[63]。而Carlson 探讨了每个基元模式的成本效益与大肠杆菌复杂行为的关系[64]。这里的成本效益主要是指生成1摩尔生物质消耗的葡萄糖(电子供体)和生成1摩尔生物质消耗的氧(电子配体)。Wessely 等组合基元通量趋势、网络推断和动态优化等方法,集成基因组和蛋白质组等数据,发现大肠杆菌的转录调控与每条基元模式上的蛋白投资相关并且存在不同的调控机制[65]。集合基因序列信息和基元模式,莱茵衣藻氮代谢的生理周期代谢调控机制最近也取得了进展[66]。探索代谢调控机理,综合反映基因和环境扰动对代谢网络的影响,也可以为菌株和藻株的突变体设计提供参考。
2.6 菌株设计
突变体构建的策略包括敲除副产物的合成基因、构建新的代谢途径、调控代谢通量在关键节点的分布等。可以通过分子生物学手段实现单基因敲除、多基因敲除、基因的过量表达等。以往的研究通常把代谢途径上的第一个酶作为关键或限速步骤,比如过量表达微藻油脂合成中的乙酰辅酶A 羧化酶却又效果不大。基元模式分析为菌株改造提供了理论框架,利用系统生物学原理,从代谢网络整体出发,计算基因工程改造与目标代谢产物产率的关系,寻找基因工程改造的目的基因。已成功应用大肠杆菌、枯草芽胞杆菌和克雷伯氏肺炎杆菌的改造[67-73]。敲除乳酸脱氢酶和丙酮酸脱氢酶复合体的枯草芽胞杆菌突变体,其异丁醇的产率可以达到理论值的61%,是野生株的2.3倍[68];大肠杆菌多基因敲除突变体以甘油为底物,每升可产乙醇40 g,是理论值的90%[70];大肠杆菌多基因敲除突变体的类胡萝卜素产率是野生株的4倍[71]。这些实例都说明,基元模式菌株设计是有效的(详见表2)。
基于基元模式分析的菌株设计策略有两类。一类是先构筑代谢网络模型,计算基元模式;再对基元模式进行分类,保留能够进行生物质合成同时生产目标产物的基元模式,敲除其他副产物的基元模式。如果代谢网络模型很大,代谢产物很多,按照这种方法需要敲除的基因过多。而且一般来说,这类策略敲除的是代谢副产物合成基因。另一类就不仅仅关注副产物的合成基因。通过分析每条基元模式的目标产物产率,以提高目标产物整体产率和保证生物质合成为目标,在全网络范围内搜索目的基因。文献[74]提出了一种基于整数算法的优化策略,敲除目的基因提高大肠杆菌生产乙醇的产率(5010个基元模式)。Flux Design 通过统计关联,寻找目的代谢产物高表达的过量表达或敲除基因,并以谷氨酸棒杆菌(60个反应)和黑曲霉(220个反应)为例分别考察了赖氨酸和酶的生产[75]。CASOP 通过基元模式评价反应重要性来选择过量表达和敲除的目的基因来提高产率,该文以107个反应的大肠杆菌模型为例考察了琥珀酸的过量生产[76]。Flux Design 和CASOP 仅能提供定性的描述,而本文作者开发的基因改变通量算法可以定量地描述基因过量表达、部分或完全失活后的代谢通量分布[49]。优化菌株设计准则,提高突变体代谢通量的定量预测能力,是下一步要研究的内容。建议综合多个软件或算法的优化结果选择改造的候选目的基因。
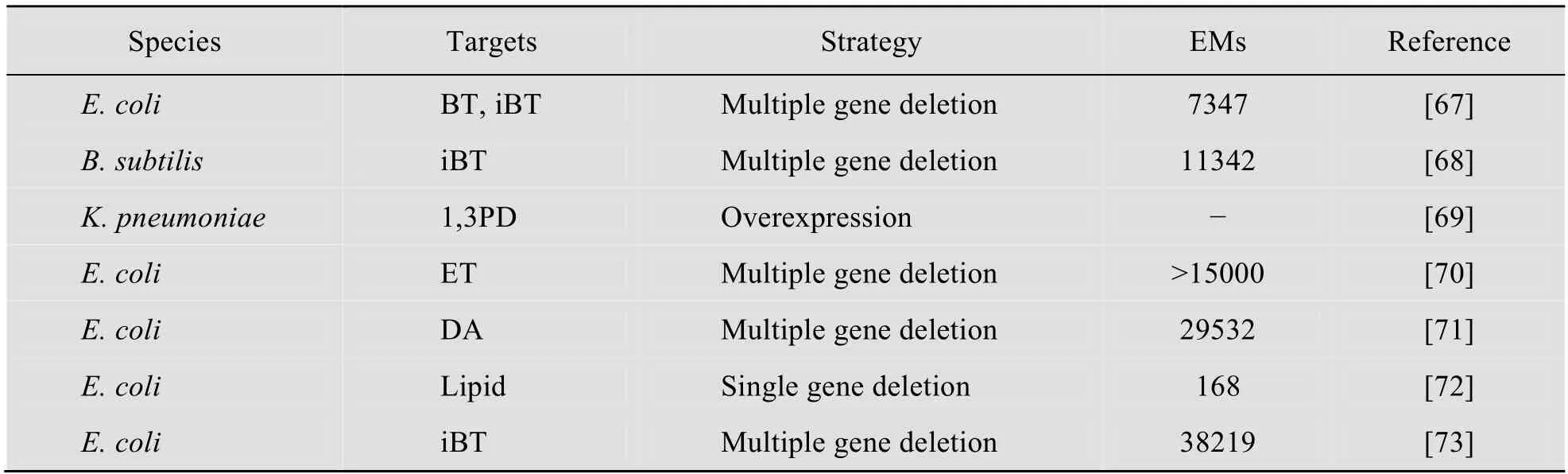
表2 基元模式分析菌株设计实例Table 2 Examples of Strain Design by EMA
2.7 信号传导网络
最初,基元模式和极端途径主要用于代谢网络的分析,并且有了广泛的应用。将代谢网络的分析方法拓展到基因调控网络和信号传导网络,也是系统生物学的最新进展。如果用有向图来描述基因调控网络,其节点是基因,而边是基因间的相互作用;对信号传导网络,节点是信号分子,而边是布尔逻辑关系,表达激活或抑制;代谢网络中的节点是代谢物,而边是生化反应。代谢网络的显著特点是质量平衡,而信号传导是信息的流动。在CellNetAnzlyzer 中,就可以构建和分析T 细胞的信号传导网络[18,22]。信号传导网络较代谢网络复杂,还需要设计新的算法计算信息流分布,并集成信号传导网络和代谢网络系统分析信号传导与代谢的关系。
3 结论与展望
基元模式已经成为系统生物学研究的重要平台工具。研究对象从代谢网络发展到信号传导网络;研究尺度从基因到生物过程以及生态系统;数学描述从稳态分解到动态解析;研究领域从微生物代谢到人类疾病。
基元模式分析的基础是准确、完备的代谢网络信息。代谢网络模型越充实,相对而言可以更准确的描述生命系统,基元模式的组合爆炸问题也会更突出,这也是限制基元模式分析应用的主要因素。通过改进算法可以提高计算速度,但分析这些基元模式仍是困难的。根据研究体系的需要,选择适当规模的代谢网络模型,或者根据添加限制条件,选择最关键的基元模式加以分析是较为可行的方法。以基元模式为基础,结合组学数据和热力学等限制条件,可以深入理解代谢调控规律和构建新的代谢途径。基于基元模式的菌株设计算法取得了一定进展。集成野生株实验数据可以简化代谢网络结构,更有针对性地寻找基因工程改造的目的基因,提高菌株设计效率。相信基元模式分析在生物网络和途径分析中将会有更多的应用。
[1]Hao T,Ma HW,Zhao XM.Progress in automatic reconstruction and analysis tools of genome-scale metabolic network.Chin J Biotech,2012,28(6):661−670(in Chinese).郝彤,马红武,赵学明.基因组尺度代谢网络自动重构及分析工具研究进展.生物工程学报,2012,28(6):661−670.
[2]Wang H,Ma HW,Zhao XM.Progress in genome-scale metabolic network:a review.Chin J Biotech,2010,26(10):1340−1348(in Chinese).王晖,马红武,赵学明.基因组尺度代谢网络研究进展.生物工程学报,2010,26(10):1340−1348.
[3]Liu LM,Chen J.Reconstruction and application of genome-scale metabolic network model.Chin J Biotech 2010,26(9):1176−1186(in Chinese).刘立明,陈坚.基因组规模代谢网络模型构建及其应用.生物工程学报,2010,26(9):1176−1186.
[4]Llaneras F,Pico J.Which metabolic pathways generate and characterize the flux space? A comparison among elementary modes,extreme pathways and minimal generators.J Biomed Biotechnol,2010,753904.
[5]Trinh CT,Wlaschin A,Srienc F.Elementary mode analysis:a useful metabolic pathway analysis tool for characterizing cellular metabolism.Appl Microbiol Biotechnol,2009,81(5):813−826.
[6]Ip K,Colijn C,Lun DS.Analysis of complex metabolic behavior through pathway decomposition.BMC Syst Biol,2011,5:91.
[7]Terzer M,Stelling J.Large-scale computation of elementary flux modes with bit pattern trees.Bioinformatics,2008,24(19):2229−2235.
[8]Jevremovic D,Trinh CT,Srienc F,et al.Parallelization of nullspace algorithm for the computation of metabolic pathways.Parallel Comput,2011,37(6/7):261−278.
[9]Acuna V,Chierichetti F,Lacroix V,et al.Modes and cuts in metabolic networks:Complexity and algorithms.Biosystems,2009,95(1):51−60.
[10]Acuna V,Marchetti-Spaccamela A,Sagot MF,et al.A note on the complexity of finding and enumerating elementary modes.Biosystems,2010,99(3):210−214.
[11]Schwartz JM,Gaugain C,Nacher JC,et al.Observing metabolic functions at the genome scale.Genome Biol,2007,8(6):R123.
[12]Kaleta C,de Figueiredo LF,Schuster S.Can the whole be less than the sum of its parts? Pathway analysis in genome-scale metabolic networks using elementary flux patterns.Genome Res,2009,19(10):1872−1883.
[13]de Figueiredo LF,Podhorski A,Rubio A,et al.Computing the shortest elementary flux modes in genome-scale metabolic networks.Bioinformatics,2009,25(23):3158−3165.
[14]Song HS,Ramkrishna D.Reduction of a set of elementary modes using yield analysis.Biotechnol Bioeng,2009,102(2):554−568.
[15]Rezola A,de Figueiredo LF,Brock M,et al.Exploring metabolic pathways in genome-scale networks via generating flux modes.Bioinformatics,2011,27(4):534−540.
[16]Peres S,Vallee F,Beurton-Aimar M,et al.ACoM:A classification method for elementary flux modes based on motif finding.Biosystems,2011,103(3):410−419.
[17]Ferreira AR,Dias JM,Teixeira AP,et al.Projection to latent pathways (PLP):a constrained projection to latent variables (PLS) method for elementary flux modes discrimination.BMC Syst Biol,2011,5:181.
[18]Klamt S,Saez-Rodriguez J,Gilles ED.Structural and functional analysis of cellular networks with CellNetAnalyzer.BMC Syst Biol,2007,1:2.
[19]Rocha I,Maia P,Evangelista P,et al.OptFlux:an open-source software platform for in silico metabolic engineering.BMC Syst Biol,2010,4:45.
[20]Schwarz R,Liang C,Kaleta C,et al.Integrated network reconstruction,visualization and analysis using YANAsquare.BMC Bioinformatics,2007,8:313.
[21]Xavier D,Vazquez S,Higuera C,et al.Tools-4-Metatool (T4M):online suite of web-tools to process stoichiometric network analysis data from Metatool.Biosystems,2011,105(2):169-172.
[22]Klamt S,von Kamp A.An application programming interface for CellNetAnalyzer.Biosystems,2011,105(2):162−168.
[23]Lewis NE,Nagarajan H,Palsson BO.Constraining the metabolic genotype-phenotype relationship using a phylogeny of in silico methods.Nat Rev Microbiol,2012,10(4):291−305.
[24]Klamt S,Grammel H,Straube R,et al.Modeling the electron transport chain of purple non-sulfur bacteria.Mol Syst Biol,2008,4:156.
[25]de Figueiredo LF,Schuster S,Kaleta C,et al.Can sugars be produced from fatty acids? A test case for pathway analysis tools.Bioinformatics,2008,24(22):2615−2621.
[26]Zhang Q,Xiu Z.Metabolic pathway analysis of glycerol metabolism in Klebsiella pneumoniae incorporating oxygen regulatory system.Biotechnol Prog,2009,25(1):103−115.
[27]Taffs R,Aston JE,Brileya K,et al.In silico approaches to study mass and energy flows in microbial consortia:a syntrophic case study.BMC Syst Biol,2009,3:114.
[28]Teusink B,Wiersma A,Jacobs L,et al.Understanding the adaptive growth strategy of Lactobacillus plantarum by in silico optimisation.PLoS Comput Biol,2009,5(6):e1000410.
[29]Xiong W,Gao C,Yan D,et al.Double CO2fixation in photosynthesis-fermentation model enhances algal lipid synthesis for biodiesel production.Bioresour Technol,2010,101(7):2287−2293.
[30]Rugen M,Bockmayr A,Legrand J,et al.Network reduction in metabolic pathway analysis:elucidation of the key pathways involved in the photoautotrophic growth of the green alga Chlamydomonas reinhardtii.Metab Eng,2012,14(4):458−467.
[31]Larhlimi A,Blachon S,Selbig J,et al.Robustness of metabolic networks:a review of existing definitions.Biosystems,2011,106(1):1−8.
[32]Behre J,Wilhelm T,von Kamp A,et al.Structural robustness of metabolic networks with respect to multiple knockouts.J Theor Biol,2008,252(3):433−441.
[33]Min Y,Jin X,Chen M,et al.Pathway knockout and redundancy in metabolic networks.J Theor Biol,2011,270(1):63−69.
[34]Haus UU,Klamt S,Stephen T.Computing knock-out strategies in metabolic networks.J Comput Biol,2008,15(3):259−268.
[35]Ballerstein K,von Kamp A,Klamt S,et al.Minimal cut sets in a metabolic network are elementary modes in a dual network.Bioinformatics,2012,28(3):381−387.
[36]Piedrafita G,Montero F,Moran F,et al.A simple self-maintaining metabolic system:robustness,autocatalysis,bistability.PLoS Comput Biol,2010,6(8):e1000872.
[37]Zhao QY,Kurata H.Maximum entropy decomposition of flux distribution at steady state to elementary modes.J Biosci Bioeng,2009,107(1):84−89.
[38]Orman MA,Berthiaume F,Androulakis IP,et al.Pathway analysis of liver metabolism under stressed condition.J Theor Biol,2011,272(1):131−140.
[39]Chan SHJ,Ji P.Decomposing flux distributions into elementary flux modes in genome-scale metabolic networks.Bioinformatics,2011,27(16):2256−2262.
[40]Orman MA,Androulakis IP,Berthiaume F,et al.Metabolic network analysis of perfused livers under fed and fasted states:incorporating thermodynamic and futile-cycle-associated regulatory constraints.J Theor Biol,2012,293:101−110.
[41]Jol SJ,Kummel A,Terzer M,et al.System-level insights into yeast metabolism by thermodynamic analysis of elementary flux modes.PLoS Comput Biol,2012,8(3):e1002415.
[42]Wlaschin AP,Trinh CT,Carlson R,et al.The fractional contributions of elementary modes to the metabolism of Escherichia coli and their estimation from reaction entropies.Metab Eng,2006,8(4):338−352.
[43]Srienc F,Unrean P.A statistical thermodynamical interpretation of metabolism.Entropy,2010,12(8):1921−1935.
[44]Unrean P,Srienc F.Metabolic networks evolve towards states of maximum entropy production.Metab Eng,2011,13(6):666−673.
[45]Unrean P,Srienc F.Predicting the adaptive evolution of Thermoanaerobacterium saccharolyticum.J Biotechnol,2012,158(4):259−266.
[46]Zhao QY,Kurata H.Estimation of intracellular flux distribution under underdetermined and uncertain conditions by maximum entropy principle.Chin J Biotech,2008,24(12):2135−2136(in Chinese).赵权宇,仓田博之.欠定和不确定条件下胞内代谢通量的最大熵原理估算.生物工程学报,2008,24(12):2135−2136.
[47]Zhao QY,Kurata H.Use of maximum entropy principle with Lagrange multipliers extends the feasibility of elementary mode analysis.J Biosci Bioeng,2010,110(2):254−261.
[48]Kurata H,Zhao Q,Okuda R,et al.Integration of enzyme activities into metabolic flux distributions by elementary mode analysis.BMC Syst Biol,2007,1:31.
[49]Zhao Q,Kurata H.Genetic modification of flux for flux prediction of mutants.Bioinformatics 2009,25(13):1702−1708.
[50]Rohwer JM.Kinetic modelling of plant metabolic pathways.J Exp Bot,2012,63(6):2275−2292.
[51]Gao J,Gorenflo VM,Scharer JM,et al.Dynamic metabolic modeling for a MAB bioprocess.Biotechnol Prog,2007,23(1):168−181.
[52]Dorka P,Fischer C,Budman H,et al.Metabolic flux-based modeling of mAb production during batch and fed-batch operations.Bioprocess Biosyst Eng,2008,32(2):183−196.
[53]Teixeira AP,Alves C,Alves PM,et al.Hybrid elementary flux analysis/nonparametric modeling:application for bioprocess control.BMC Bioinformatics,2007,8:30.
[54]Kim JI,Varner JD,Ramkrishna D.A hybrid model of anaerobic E.coli GJT001:combination of elementary flux modes and cybernetic variables.Biotechnol Prog,2008,24(5):993−1006.
[55]Young JD,Henne KL,Morgan JA,et al.Integrating cybernetic modeling with pathway analysis provides a dynamic,systems-level description of metabolic control.Biotechnol Bioeng,2008,100(3):542−559.
[56]Song HS,Morgan JA,Ramkrishna D.Systematic development of hybrid cybernetic models:Application to recombinant yeast co-consuming glucose and xylose.Biotechnol Bioeng,2009,103(5):984−1002.
[57]Song HS,Ramkrishna D.Prediction of metabolic function from limited data:Lumped hybrid cybernetic modeling (L-HCM).Biotechnol Bioeng,2010,106(2):271−284.
[58]Song HS,Ramkrishna D.Cybernetic models based on lumped elementary modes accurately predict strain-specific metabolic function.Biotechnol Bioeng,2011,108(1):127−140.
[59]Franz A,Song HS,Ramkrishna D,et al.Experimental and theoretical analysis of poly(beta-hydroxybutyrate)formation and consumption in Ralstonia eutropha.Biochem Eng J,2011,55(1):49−58.
[60]Cakir T,Kirdar B,Onsan ZI,et al.Effect of carbon source perturbations on transcriptional regulation of metabolic fluxes in Saccharomyces cerevisiae.BMC Syst Biol,2007,1:18.
[61]Schuster S,Pfeiffer T,Fell DA.Is maximization of molar yield in metabolic networks favoured by evolution? J Theor Biol,2008,252(3):497−504.
[62]Bordel S,Nielsen J.Identification of flux control in metabolic networks using non-equilibrium thermodynamics.Metab Eng,2010,12(4):369−377.
[63]Teixeira AP,Dias JML,Carinhas N,et al.Cell functional enviromics:unravelling the function of environmental factors.BMC Syst Biol,2011,5:92.
[64]Carlson RP.Decomposition of complex microbial behaviors into resource-based stress responses.Bioinformatics,2009,25(1):90−97.
[65]Wessely F,Bartl M,Guthke R,et al.Optimal regulatory strategies for metabolic pathways in Escherichia coli depending on protein costs.Mol Syst Biol,2011,7:515.
[66]Schauble S,Heiland I,Voytsekh O,et al.Predicting the physiological role of circadian metabolic regulation in the green alga Chlamydomonas reinhardtii.PLoS ONE,2011,6(8):e23026.
[67]Trinh CT.Elucidating and reprogramming Escherichia coli metabolisms for obligate anaerobic n-butanol and isobutanol production.Appl Microbiol Biotechnol,2012,95(4):1083−1094.
[68]Li S,Huang D,Li Y,et al.Rational improvement of the engineered isobutanol-producing Bacillus subtilis by elementary mode analysis.Microb Cell Fact,2012,11(1):101.
[69]Chen Z,Liu H,Liu D.Metabolic pathway analysis of 1,3-propanediol production with a genetically modified Klebsiella pneumoniae by overexpressing an endogenous NADPH-dependent alcohol dehydrogenase.Biochem Eng J,2011,54(3):151−157.
[70]Trinh CT,Srienc F.Metabolic engineering of Escherichia coli for efficient conversion of glycerol to ethanol.Appl Environ Microbiol,2009,75(21):6696−6705.
[71]Unrean P,Trinh CT,Srienc F.Rational design and construction of an efficient E.coli for production of diapolycopendioic acid.Metab Eng,2010,12(2):112−122.
[72]Kenanov D,Kaleta C,Petzold A,et al.Theoretical study of lipid bio-synthesis in wild-type Escherichia coli and in a protoplast-type L-form using elementary flux mode analysis.FEBS J,2010,277(4):1023−1034
[73]Trinh CT,Li J,Blanch HW,et al.Redesigning Escherichia coli metabolism for anaerobic production of isobutanol.Appl Environ Microbiol,2011,77(14):4894−4904.
[74]Jungreuthmayer C,Zanghellini J.Designing optimal cell factories:integer programing couples elementary mode analysis with regulation.BMC Syst Biol,2012,6:103.
[75]Melzer G,Esfandabadi ME,Franco-Lara E,et al.Flux design:in silico design of cell factories based on correlation of pathway fluxes to desired properties.BMC Syst Biol,2009,3:120.
[76]Haedicke O,Klamt S.CASOP:A computational approach for strain optimization aiming at high productivity.J Biotechnol,2010,147(2):88−101.
猜你喜欢
计算机工程与应用(2023年1期)2023-01-13
农业灾害研究(2022年1期)2022-05-07
中学生数理化·七年级数学人教版(2019年10期)2019-11-25
中学生数理化·高一版(2018年9期)2018-10-09
湖南教育·C版(2018年3期)2018-06-05
科学导报(2018年30期)2018-05-14
化工进展(2015年6期)2015-11-13
中国海洋大学学报(自然科学版)(2014年12期)2014-02-28
中国土地科学(2011年11期)2011-03-20
