
1,4-二苄基-1H-咪唑-5-氧三氟甲磺酸的合成*
2013-09-01 02:12王月慧严生虎沈介发刘建武
合成化学 2013年6期
吴 菊,马 兵,王月慧,张 跃,严生虎,沈介发,刘建武
(常州大学制药与生命科学学院,江苏 常州 213164)
天然产物Theonella家族的系列化合物具有类似的双环十二肽结构。其中包括具有偶联结构的 Theonellamide F[1](Ⅰ,Chart 1)和 Theopalauamide[2](Ⅱ,Chart 1)。Ⅰ和Ⅱ都包含八个非天然的氨基酸,同时还包括一个奇特的τ-L-组氨酸-D-丙氨酸(τ-L-His-D-Ala)的双氨基酸偶联结构,通过咪唑环τ-N与丙氨酸β-C连接。遗憾的是,到目前为止还未有Ⅰ的全合成报道。而且,只有两个课题组曾在这方面做过一定的尝试[3]。

Chart 1
20世纪90年代,Hamada和Shioiri曾报道Ⅰ中的关键片段 Aboa[4]和 Ahad[5],同时也分别对它的左手环[6]和右手环[7]的合成进行构建。而对于τ-L-His-D-Ala的合成,尽管Hamada和Shioiri提出了一条相对简洁的路径(采用β-内酯开环与组氨酸直接偶联的方式进行合成[8]),但是此方法的低区域选择性以及需要消耗大过量的组氨酸原料,导致总收率低(7%)。而在此后的十六年间却没有相关的全合成进展。这很有可能是由于中间体τ-L-His-D-Ala低效率的合成导致无法积累足够的量来完成整个分子的合成。因此,τ-L-His-D-Ala的成功合成将成为Theonella化合物全合成的关键步骤。
本文设计一条收敛的合成路线(Scheme 1),期望能解决Hamada合成中低收率和低区域选择性问题。在Scheme 1的逆合成分析中,最重要的是实现咪唑环合成,主要通过3直接环合。而3则可以通过4和5来合成。
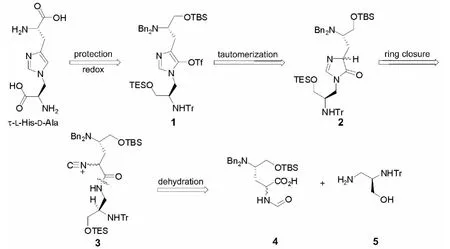
Scheme 1
4可从D-丝氨酸出发,经8步反应合成。5可从L-丝氨酸出发,经3步反应合成,收率70%[9]。但是,现有的实验经验使我们认识到,环合不一定能成功,即2不确定能否成功合成。为此,我们设计了并合成了一个简单的化合物——1,4-二苄基-1H-咪唑-5-甲基三氟甲磺酸酯(6)来作为τ-LHis-D-Ala的模型构建环合过程,通过其合成研究探索一种τ-L-His-D-Ala的全新合成方法。
通过6的逆合成分析可知,合成6得先合成N-苄基-2-异氰基-3-苯基丙酰胺(7)。为此,本文以苯丙氨酸(8)为起始原料,经5步反应合成了7;7经环合合成 6(Scheme 2),其结构经1H NMR,13C NMR,IR和MS确证。并重点考察了碱和氟化剂对环合反应的影响,得出环合反应的最佳条件。
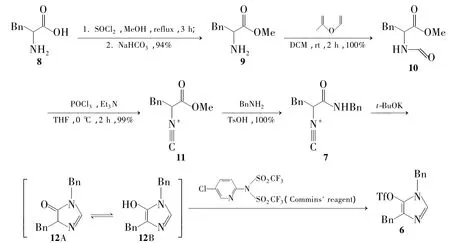
Scheme 2
6的合成,成功验证了从α-异氰基酰胺直接成环合成成咪唑环的可行性。
1 实验部分
1.1 仪器与试剂
Bruker AVANCEⅢ500 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);FT-IR-8400S型红外光谱仪(KBr压片);Finnigan TSQ Quantum-MS型质谱仪。
柱层析用硅胶200目~300目,青岛海洋化工厂;其余所用试剂均为分析纯。
1.2 合成
(1)苯丙氨酸甲酯(9)的合成
在反应瓶中加入8 16.52 g(100 mmol)和无水甲醇30 mL,搅拌得悬浮液;Ar气氛保护下于0℃滴加亚硫酰氯18.00 mL(253 mmol),滴毕,慢慢升至室温,回流反应4 h。蒸除溶剂得甲基酯盐酸盐。用饱和NaHCO3溶液(100 mL)洗涤,用二氯甲烷(4×50 mL)萃取,合并有机层,用无水MgSO4干燥,浓缩得无色油状物 9 16.83 g,收率94%;1H NMR δ:7.30~7.10(m,5H),3.72~3.65(m,1H),3.69(s,3H),3.00~ 2.80(m,2H),1.49(brs,2H)。
(2)N-甲酰基-苯丙氨酸甲酯(10)的合成
在反应瓶中加入9 8.95 g(50 mmol)和无水二氯甲烷13 mL,搅拌使其溶解;Ar气氛保护下于0℃滴加混合酸酐[V(乙酸酐)∶V(甲酸)=1∶1]6.72 g(80 mmol),滴毕,于室温反应2 h。蒸除溶剂得淡黄色油状物10,收率100%;1H NMR δ:8.01(s,1H),7.29~7.13(m,7H),6.88(brs,1H),3.66(dd,J=8.0 Hz,6.0 Hz,1H),3.64(s,3H),3.02(dd,J=7.8 Hz,6.0 Hz,1H)。
(3)异腈化物(11)的合成
在反应瓶中加入三氯氧磷2.9 mL(31 mmol)和THF 60 mL,搅拌使其溶解;于-78℃滴加混合液[三乙胺17 mL+10 5.18 g(25 mmol)+THF 60 mL],滴毕,于0℃反应直至反应液变为红色为止(约2 h)。加水50 mL,用乙醚(3×30 mL)萃取,合并有机层,用MgSO4干燥,浓缩得红色油状物 11;1H NMR δ:7.29~7.17(m,5H),4.39(dd,J=8.4 Hz,4.7 Hz,1H),3.73(s,3H),3.19(dd,J=13.8 Hz,4.7 Hz,1H),3.07(dd,J=13.8 Hz,8.4 Hz,1H)。
(4)7的合成
在反应瓶中依次加入 11 5.95 g(31.5 mmol),苄胺 3.38 g(31.5 mmol)及 p-甲苯磺酸(TsOH)31.5 mg(0.183 mmol),搅拌下于60 ℃反应35 min。Ar气氛下,有灰白色沉淀析出。过滤,滤饼用甲醇洗涤,异丙醇重结晶得象牙色粉末7 8.42 g,收率 100%;1H NMR δ:7.34~7.29(m,5H),7.27(s,1H),7.25(s,1H),7.14(s,1H),7.13(s,1H),6.49(br s,1H),4.50~4.45(m,3H),4.39(dd,J=15.0 Hz,5.0 Hz,1H),3.31(dd,J=15.0 Hz,5.0 Hz,1H),3.22(dd,J=15.0 Hz,5.0 Hz,1H);13C NMR δ:164.5,162.4,137.5,134.3,129.6(2C),128.8,128.7,127.9(2C),127.8(2C),127.7(2C),59.8,44.0,38.6;IR ν:3 298,3 064,3 030,2 930,2 142,1 666,1 604,1 534,1 496,1 454,1 286,1 240,1 081,1 029 cm-1;HR-MS(ESI)m/z:Calcd for C17H16N2OH[MH+]265.136 1,found 264.321 7。
(5)6的合成
Ar保护,在反应瓶中加入干燥 THF 26.8 mL,搅拌下于 -78 ℃ 加入 7 709.2 mg(2.68 mmol);缓慢滴加叔丁醇钾2.68 mL(1 mol·L-1的THF溶液),滴毕,于 -20℃反应20 min;于-78℃缓慢加入 Commins’试剂 2.1 g(5.37 mmol)的THF(1.8 mL)溶液,反应1 h;于室温反应过夜。旋干溶剂,用二氯甲烷(3×30 mL)萃取,合并有机层,用水洗涤,无水Na2SO3干燥,浓缩后经硅胶柱层析[洗脱剂:V(正己烷)∶V(乙酸乙酯)=95∶5]纯化得淡黄色油状物 6,收率30%;1H NMR δ:7.39~7.28(m,6H),7.24~7.71(m,3H),7.13~ 7.06(m,2H),5.56(s,2H),4.05(s,2H);13C NMR δ:136.5,135.0,133.2,139.2,129.0(2C),128.6,128.5,127.9(2C),127.3(2C),126.9(2C),126.6(2C),49.5,32.2;HR-MS(ESI)m/z:Calcd for C18H15N2O3SF3[MH+]397.087 7,found 397.087 7。
2 结果与讨论
2.1 7 的合成
对于7的合成,文献[10]方法是以外消旋8,亚硫酰氯和甲醇进行甲酯化得中间体9。乙酸酐和甲酸配成混合酸酐与9反应生成10。通常,甲酰胺到异氰酸的转化,主要是利用Burgess试剂进行脱水,目的是为了避免与分子中其他的氮原子发生潜在的副反应。但在10中,本文仅利用磷酰氯脱水重结晶后,得到几乎等当量的11;11再通过TsOH催化成功转化为7,并直接从反应混合物中析出。
2.2 6 的合成
7的环合过程是本文的关键。我们考察了不同类型的碱对7环合反应的影响,结果见表1。从表1可见,可能是生成了不稳定的互变异构体12,从而导致环合效果不佳,即收率低。众所周知,12在碱性条件下会发生两种构型异构体的转变。当然,得到烯醇式化合物12B将是有利于此反应的。与此同时,采用三氟甲基磺酸试剂将其转变为相应的三氟甲磺酸酯,可稳定此异构体。
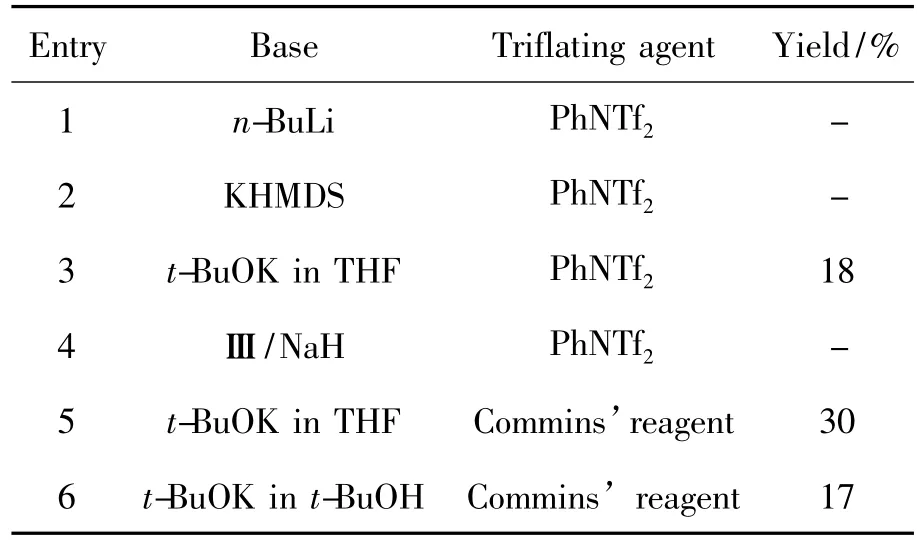
表1 咪唑环合成条件筛选Table 1 Screening of imidazole ring synthesis conditions
以n-BuLi为碱,PhNTf2(Chart 2)为磺酸酯化剂时,随着反应的进行,实验中未能检测到6的生成(Entry 1)。尝试了KHMDS(Chart 2),t-BuOK和膦碱(Ⅲ,Chart 2)分别与PhNTf2进行的反应。结果表明,以t-BuOK为碱时,可以得18%的6(Entry 3),而在其它两种条件下只能得到一些未知的混合物,并没有6生成(Entry 2和Entry 4)。
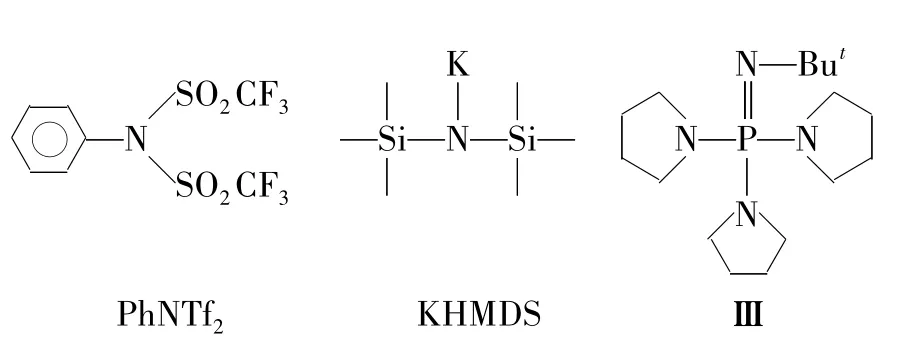
Chart 2
对三氟甲基磺酸酯试剂进行了摸索,采用较为经典的Commins’试剂作为磺酰化试剂,发现有产物生成。同时,我们对反应条件进行适当的优化,产品收率能提高至30%(Entry 5),但6在柱层析纯化过程中,多余的磺酸酯试剂总会与所需产物一同洗脱下来,而无法得到纯度较高的6(不影响收率的计算,收率是通过总重量和核磁共振摩尔百分比准确计算的)。
综上所述,合成6的最佳碱为t-BuOK,最佳酯化剂为 Commins’reagent。
3 结论
通过建立τ-L-His-D-Ala模型研究已经成功地证实了咪唑环环合的可行性,同时初步筛选出合适的碱及三氟甲磺酸酯化试剂。从4和5的偶联后生成二肽异氰类化合物的合成已经同时展开,并且已经合成出化合物3[11]。模型化合物6的合成经验,将很快用于实现从3到1的转变,并最终完成高选择性的二肽τ-L-His-D-Ala合成。
本文提出了一种新型1,4-二取代咪唑环的合成方法,即通过α-异氰基酰胺直接成环合成咪唑环。模型化合物1,4-二苄基-1H-咪唑-5-甲基三氟甲磺酸酯的合成,将帮助解决二肽τ-L-His-DAla合成中区域选择性问题。
[1] Matsunaga S,Fusetani N,Hashimoto K,et al.A novel antifungal bicyclic peptide from a marine sponge Theonella Sp[J].J Am Chem Soc,1989,(111):2582-2588.
[2] Schmidt E W,Bewley C A,Faulkner D J.Theopalauamide,a bicyclic glycopeptide from filamentous bacterial symbionts of the lithistid sponge Theonella swinhoei from palau and mozambique[J].J Org Chem,1998,63:1254-1258.
[3] Schmidt E W,Obraztsova A Y,Davidson S K,et al.Identification of the antifungal peptide-containing symbiont of the marine sponge Theonella swinhoei as a novel proteobacterium,candidatus entotheonella palauensis[J].Mar Biol,2000,1:337-341.
[4] Tohdo K,Hamada Y,Shioiri T.Theonellamide F synthetic studies.Stereoselective synthesis of(3S,4S,5E,7E)-3-amino-8-(4-bromophenyl)-4-hydroxy-6-methyl-5,7-octadienoic acid(aboa)[J].Tetrahedron Lett,1992,33:2031-2034.
[5] Tohdo K,Hamada Y,Shioiri T.Stereoselective synthesis of(2S,4R)-2-amino-4-hydroxyadipic acid,the characteristic amino acid of theonellamide F[J].Synlett,1994:105-106.
[6] Tohdo K,Hamada Y,Shioiri T.Synthesis of the southern hemisphere of theonellamide F,a bicyclic dodecapeptide of marine origin[J].Synlett,1994:247-249.
[7] Tohdo K,Hamada Y,Shioiri T.Synthesis of the northern hemisphere of theonellamide F,a bicyclic dodecapeptide of marine origin[J].Synlett,1994:250.
[8] Arnold L D,Kalantar T H,Vederas J C.Conversion of serine to stereochemically pure β-substituted α-amino acids via β-lactones[J].J Am Chem Soc,1985,(107):7105-7109.
[9] Pickersgill I F,Rapoport H.Preparation of functionalized,conformationally constrained DTPA analogues from L-or D-serine and trans-4-hydroxy-L-proline.Hydroxymethyl substituents on the central acetic acid and on the backbone[J].J Org Chem,2000,65:4048-4057.
[10] Elders N,Schmitz R F,de Kanter F J,et al.A resource-efficient and highly flexible procedure for a three-component synthesis of 2-imidazolines[J].J Org Chem,2007,72:6135-6142.
[11] 王月慧,马兵,吴菊,等.二肽物±(4S)-5-[叔丁基二甲基硅氧-4-二苄氨基-2-甲酰氨基-N-3-羟基-2-(三苯甲烷基)丙基]戊酰胺的合成[J].合成化学,2013,21(1):11-15.
猜你喜欢
海洋通报(2022年6期)2023-01-07
海南大学学报(自然科学版)(2020年2期)2020-07-21
有机氟工业(2020年2期)2020-07-04
分析化学(2017年12期)2017-12-25
发酵科技通讯(2016年4期)2016-12-21
合成化学(2015年10期)2016-01-17
合成化学(2015年10期)2016-01-17
合成化学(2015年1期)2016-01-17
中国洗涤用品工业(2015年9期)2015-02-28
有机氟工业(2014年3期)2014-06-05
