
放射性核素铀在针铁矿中的占位研究*
2013-09-27 11:03金宝蔡军陈义学
物理学报 2013年8期
金宝 蔡军 陈义学
(华北电力大学核科学与工程学院,北京 102206)
(2012年8月6日收到;2012年12月24日收到修改稿)
1 引言
随着核废料处理压力越来越大,人们更加关注放射性核素铀在土壤中的吸附特征.针铁矿是土壤中分布广泛的一种重要物质,它对重金属具有强烈的亲和作用[1-3].针铁矿(α-FeOOH)属于Pnma正交晶体结构,它的晶格常数为a=9.9510A˚,b=3.0178A˚,c=4.62A˚[4,5].Forsyth等[6]利用中子衍射对土壤中存在的以及人工合成的针铁矿样品进行检测,发现针铁矿在373 K以下是反铁磁性的.2008年Kubicki等[7]用密度泛函理论计算了针铁矿体相与其(010)表面的相关能量,同时对它们的结构与振动频率也进行了计算.Leung和Criscenti[3]用第一性原理研究了针铁矿表面羟基群的酸性常数性质.
最近Kerist等[8]应用经验的分子动力学方法研究单个铀原子在针铁矿中的占位性质.发现放射性同位素铀容易掺杂在针铁矿八面体的间隙位和替代位,还发现对于针铁矿中Fe原子的第一壳层的羟基脱去质子后,铀原子可以代替Fe而不会引起晶格大的畸变.而在八面体间隙位可引起7倍大的晶格形变.据我们所知,对于铀原子掺入到针铁矿后的形成能及结合能的研究以及双铀原子在针铁矿的掺入还没有见文献报道.本研究将利用第一性原理方法对单个铀原子及双铀原子掺入针铁矿的具体构型及其形成能与结合能进行更深入的研究.
2 计算方法
本研究用VASP[9-12]软件进行计算.选用广义梯度函数投影缀加波赝势[13]描述放射性核素原子在针铁矿中的占位时原子间的相互作用.交换关联函数采用广义梯度近似Perdewe-Wang 91形式[14,15].关联梯度方法被用来对原子进行弛豫.为了模拟针铁矿铁原子上的电子自旋方向沿(010)面正负交替的反铁磁性[6],电子自旋极化计算被考虑.因为铁及铀原子在针铁矿中其电子之间存在强关联作用,我们采用GGA+U方法,其中对于铀与铁原子的关联参数分别取为U=4.6,4.5 eV;J=0.5,0 eV[16-19].在目前的计算中针铁矿超晶胞由1×3×2个单胞构成,包含24个铁原子,48个氧原子,24个氢原子.为了对布里渊区求和,用Monkhorst-Pack方法[20]划分k空间网格,2×2×2个k点标样数被使用.平面波动能的截止半径为401 eV.电子自由度收敛判据是两个步长之间的总能变化小于10-5eV.离子弛豫收敛判据是两个离子步长的系统总能之差小于10-4eV.
我们计算缺陷的形成能与结合能定义为[21,22]:超级晶胞的能量与其所包含的所有原子的基态能量的和之差;如果形成能为正则其形成需要吸收能量,为负则其形成过程会放出能量;形成能大于零且越大则表示这种系统形成需要的外界能量越大,即越不容易形成,也不太稳定,反之则相反.结合能定义为[21,22]:有两个相互影响的缺陷系统的形成能与各含有单个缺陷的两个系统的形成能的和之差.结合能为正则表示两缺陷之间相互排斥,为负两缺陷相互吸引.
3 结果与讨论
针铁矿属于Pnma正交晶体结构,它的实验晶格常数为a=9.9531A˚,b=3.0178A˚,c=4.5979A˚,包括了8个氧原子,4个铁原子与4个氢原子.如果不考虑反铁磁性氧原子有两个等效位子,其实验分数坐标分别为(0.19914,0.25,0.70569)和(0.05298,0.25,0.19871),铁与氢原子各有一个等效位子,它们的实验分数坐标分别为(0.85366,0.25,0.04892)和(0.08170,0.25,0.37813).用VASP软件优化针铁矿的晶格常数与原子坐标获得理论晶格常数为a=10.0028A˚,b=3.0432A˚,c=4.6508A˚ 以及相应的理论分数坐标分别是(0.19777,0.25000,0.69889),(0.05528,0.25000,0.19709),(0.85377,0.25000,0.04911),(0.08652,0.25000,0.40003).这些理论结果都与上面的实验结果[4,5]一致.用理论晶格常数构建针铁矿的1×3×2超晶胞,包括24铁、24氢和48氧共96个原子.我们研究单个铀原子及两个铀原子在超晶胞中的多种占位特征.用O,T,S分别表示铀原子占八面体间隙位(O),四面体间隙位(T)和铀原子替代针铁矿中的Fe原子的替代位(S).SS,OS,TS分别表示一个铀原子分别为替位原子、八面体与四面体间隙原子时其最近邻存在一个替位铀原子.它们的初始构型如图1(a)—(c)所示.
图1(a)对应铀原子在针铁矿中的S位掺入,(b)与(c)分别为O和T位掺入,从图1(a)中可见铀原子在替代位掺入时,铀原子最近邻有8个铁的最近邻,平均距离为3.336˚A,有6个氧最近邻,平均距离为2.048˚A,弛豫之后铀原子与8个铁原子的平均距离,及与氧的平均距离的变化都小于0.01%,其结构非常稳定.图1(b)显示八面体间隙位的铀原子掺入,其顶点是由两个氧构成,基面由四个氧构成,其中基面上的两个氧与氢原子构成两个羟基并与另两个氧相对排列,其余的氧没有羟基,铀原子与它们的平均距离为1.804˚A,弛豫之后铀原子与这六个氧原子的平均距离变为2.405˚A,其与周围氧原子的距离增加了33.33%.图1(c)显示四面体间隙位的铀原子掺入,其顶点是一个铁,基面由没有羟基的三个氧原子构成,铀原子与它们的平均距离为1.844˚A,弛豫后铀原子与它们的平均距离变为3.711˚A,其平均距离增加了101.25%.后面计算结果也显示它们平均距离的变化大小次序与其单个铀原子在针铁矿中掺入所对应的构形的形成能大小的顺序是一致的.最近Kerist等[8]用经验的分子动力学方法研究单个铀原子在针铁矿中的占位性质.发现放射性同位素铀容易掺杂在针铁矿八面体的间隙位和替代位,还发现对于针铁矿中Fe原子的第一壳层的羟基脱去质子后,铀原子可以代替Fe而不会引起晶格大的变形.而在八面体间隙位可引起7倍大的晶格形变.他们的替代位的近邻原子位置变化结果与目前结论相一致,而八面体间隙位的位置变化比目前大出很多.他们没有发现铀原子也可以掺入到针铁矿的一种四面体间隙位中.目前的计算发现,铀原子在针铁矿四面体间隙的掺入所引起的周围原子位置变化比Kerist等[8]用经验的方法计算的铀原子八面体间隙位引起的畸变还要小许多.这可能是由于他们应用经验方法计算造成的.另外由计算我们发现铀原子在Fe的替代位的电子布居数比中性铀原子少约三个电子,表明此时代位铀原子的价态是三价.
SS,OS,TS位分别是铀在S,O与T位置时与其最近邻的S位上另一个铀原子组成.它们的初始原子结构也在图1中显示.图1(a)—(c)中黄色的铁原子被一个铀原子替代分别构成了双铀原子掺入针铁矿中,组成SS,OS,TS的初始构型.弛豫的结果显示对于SS与OS其构型保持稳定(SS构型中的U—U键距离从3.045˚A增加到3.420˚A,距离增加了12.32%,而OS构型的U—U键距离从2.675˚A增加到3.485˚A,距离增加了30.28%.对于TS构型,根据弛豫后的结构图发现其结构已经完全改变,计算显示TS构型极不稳定.
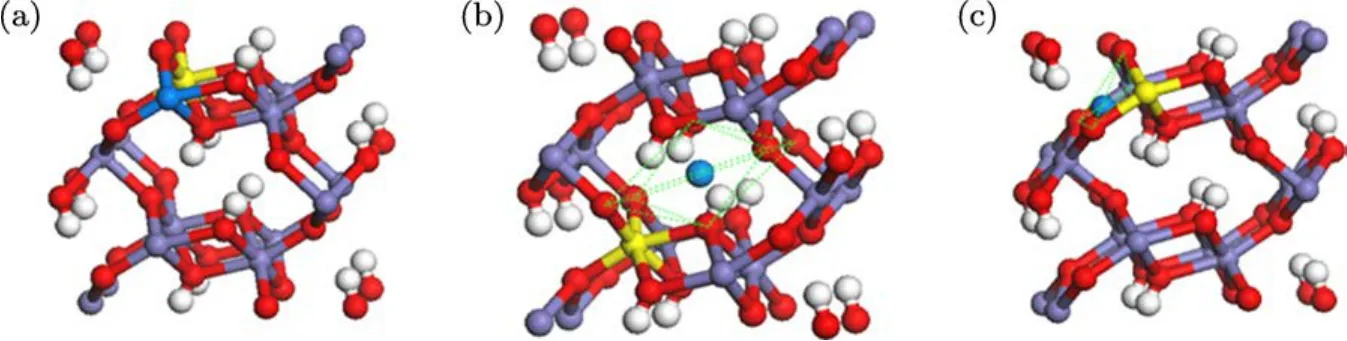
图1 S,O,T,SS,OS,TS结构图,红色为氧原子,白色为氢原子,青紫色为铁原子,蓝色为铀原子,黄色是一个Fe原子,但在双铀原子掺入的情况下它被一个铀原子替代,分别构成双铀原子掺入的SS,OS,TS结构 (a)S构型与SS构型;(b)O构型和OS构型;(c)T构型与TS构型
我们也计算了它们的形成能与结合能,结果列于表1.表中,Ef和Eb分别表示相应缺陷的形成能与结合能.其相应的初始原子构型如图1(a)—(c)所示.
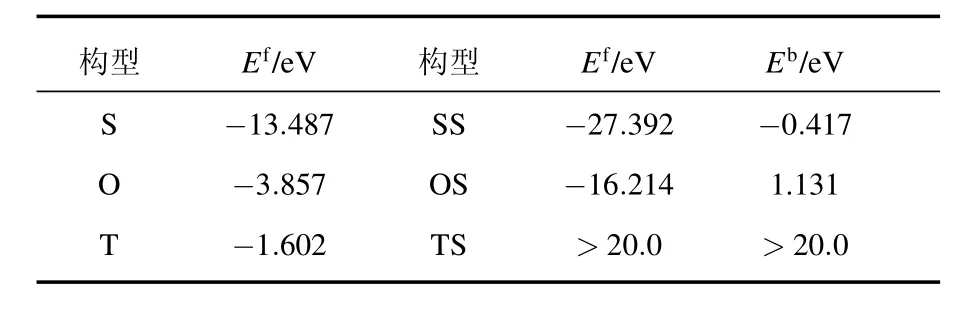
表1 铀原子及双铀原子掺杂针铁矿中的形成能(E f)与结合能(E b)
我们研究了铀替代针铁矿中的铁的替位缺陷和铀的多种八面体和多种四面体间隙缺陷,表1列出三种最稳定的单缺陷构型的形成能及三种双铀原子缺陷的形成能与其结合能.从表1可以看出,铀的替代位、一种八面体间隙位和一种四面体间隙位的形成能分别是-13.487,-3.857,-1.602 eV,而其余的八面体、四面体间隙的形成能均大于20.0 eV(表中没有显示),其结构不稳定.对于双铀原子缺陷SS位的形成能是-27.329 eV,结合能为-0.417 eV;OS位的形成能是-16.214 eV,结合能为1.131 eV.由形成能的定义知道形成能为正则其形成需要吸收能量,为负则其形成过程会放出能量,结合能为正则表示两缺陷之间相互排斥,为负两者相互吸引.单个铀原子的S位、O位、T位的形成能均小于零,说明单个铀原子与针铁矿的结合属于放热反应,其结构稳定.它们最稳定的结构为替位铀原子掺入,八面体掺入次之,四面体掺入再次之.对于双铀原子掺入到针铁矿的情形,从表1中可以看出SS位与TS位的形成能都为负,说明它们与针铁矿的反应也为放热反应,在针铁矿中极易形成.由它们的结合能的计算结果得到SS位的结合能小于零,而OS位的结合能大于零,说明这两种缺陷形成以后,两个铀原子在SS构型时相互吸引,而在OS时相互排斥,亦即两个铀原子容易以SS结构偏聚,而不易产生OS结构的偏聚.X光的实验结果显示铀原子极易掺入到针铁矿的结构当中[8],从表1可看出目前计算的S,O,T,SS,OS结构的形成能都小于零说明它们与针铁矿的反应为放热反应,它们在针铁矿中极易形成,这与X光的实验结果一致.
我们也计算了单个铀原子掺入到针铁矿中的另外一种八面体和另外6种四面体构型以及双铀原子的TT,OO构型的形成能和结合能.图2给出了它们与针铁矿中近邻原子作用的初始结构.与图1(b)周围全是氧原子不同,图2(a)对应八面体间隙位的铀原子掺入,其顶点是由两个氧原子构成,基面由两个氧和两个铁构成,其中基面上的两个氧与氢原子构成两个羟基并与铁相对排列,其余的氧没有羟基.图2(b)—(g)都对应四面体间隙位的铀原子掺入,它们的构型与图1(c)构型不同,图1(c)中顶点是一个铁原子基面是不带羟基的三个氧原子.图2(b)中的顶点是由一个氧原子构成,基面由一个氧原子两个铁原子构成;图2(c)中的顶点是由一个铁原子构成,基面由两个铁原子和一个氧原子构成;图2(d)中的顶点是由一个氧原子构成,并与一个氢原子组成一个羟基,基面由三个氧原子构成;图2(e)中的顶点是由一个氧原子构成,并与一个氢原子组成一个羟基,基面由两个氧原子和一个铁原子构成,其中一个氧原子与氢原子组成一个羟基;图2(f)中的顶点是由一个铁原子构成,基面由一个铁原子和两个氧原子构成其中一个氧原子与氢原子组成一个羟基;图2(g)的结构与图2(e)的类似,其顶点是一个铁原子,基面是三个原子,其中的两个氧原子与氢形成羟基,它们之间U—O与U—Fe的键长稍有不同;图2(h)是双铀原子占据两个相邻的八面体位(OO结构),两个八面体共用一个氧原子;图2(i)是双铀原子占据两个相邻的四面体位(TT结构),两个四面体共用一个基面.我们计算这些结构发现它们的Ef和Eb都远大于零,说明它们的结构不稳定.结构弛豫结果也表明它们的初始构型变形的十分巨大,从在其附近的原子构型已经不能辨认出针铁矿的体相结构了,这与计算能量推断的结论一致.
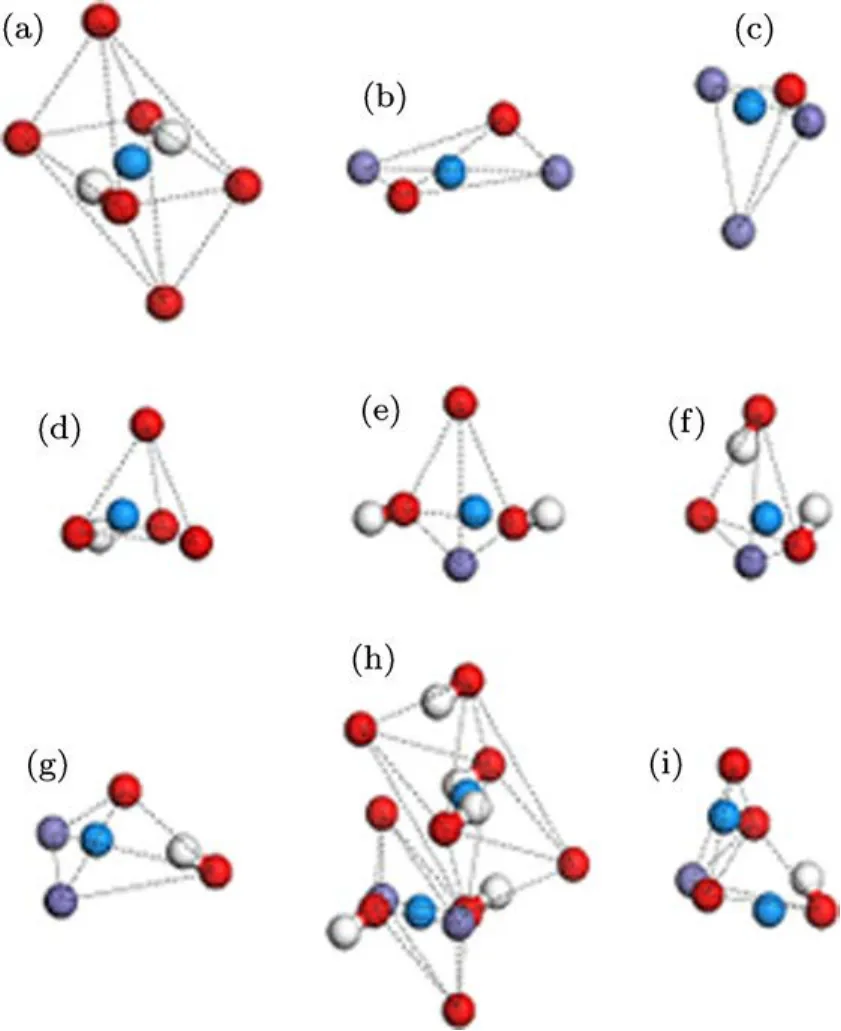
图2 铀与针铁矿中近邻原子作用的另外几种构型,红色为氧原子,白色为氢原子,青紫色为铁原子,蓝色为铀原子,OO与TT分别表示双铀原子占据两个相邻的八面体位和两个四面体位的双铀在针铁矿中的掺杂构型 (a)另一种八面体构型;(b)—(g)另外6种四面体构型;(h)OO构型;(i)TT构型
分析图1与图2的结构可以发现,图2中这些结构不稳定的原因可能是因为铀原子与其余原子之间的距离较近,空间较小所引起的.例如在八面体构型中,图1(b)中U—O距离最小为1.682˚A,而图2(a)中U—O距离最小仅为1.310˚A.并且由计算发现Fe与铀在针铁矿中同带正电荷,因此在基面上的铁原子与铀原子产生相互排斥;在四面体构型中,图1(c)中的铀原子与其余原子的初始的最小距离为1.510A˚,而图?2中6个四面体间隙位中铀原子与其余原子的初始的最小距离均小于1.304A˚.对于双铀原子占位也有同样的情形.这说明与图1比较,图2中的铀原子与周围原子距离太近导致其结构极其不稳.
4 结论
用第一性原理计算放射性核素铀在针铁矿S,O,T位的形成能,发现单个铀很容易掺入到针铁矿中的替位、八面体间隙位(其顶点是由两个氧构成,基面由四个氧构成,其中基面上的两个氧与氢原子构成两个羟基并与另两个氧相对排列,其余的氧没有羟基,弛豫后铀原子与这六个氧原子的平均距离增加了33.33%)、以及四面体间隙位的铀原子掺入(其顶点是一个铁,基面由没有羟基的三个氧原子构成,弛豫后铀原子与它们的平均距离增加了101.25%).铀原子以这样的S,O,T位掺入到针铁矿中时,其与针铁矿结合都属于放热反应.它们最稳定的结构为替位铀原子掺入,该八面体掺入次之,该四面体掺入再次之.通过计算分析双铀原子在针铁矿SS,OS,TS,OO,TT的形成能和结合能,我们发现双铀原子很容易掺入到SS和OS位,但是双原子铀在针铁矿中会以SS形式发生偏聚而难以OS偏聚.
[1]Goli E,Rahnemaie R,Hiemstra T,Malakouti M J 2011 Chemosphere 82 1475
[2]Xia SW,Zhong BW,Chou M,Yu L M 2011 Periodical of Ocean Univ.China 41 57(in Chinese)[夏树伟,钟炳伟,仇萌,于良民2011中国海洋大学学报41 57]
[3]Leung K,Criscenti L J2012 J.Phys.Condens.Matter.24 124105
[4]HayesK F,Roe A L,Brown GEB,Hodgson,K O,Leckie JO,Parks GA,1987 Science 238 783
[5]Yang H X,Lu R,Downs RT,Costin G 2006 Structure Report Online 62 i250
[6]Forsyth JB,Hedley IG,Johnson CE 1968 J.Phys.C 1 179
[7]Kubicki JD,Paul K W,Sparks D L 2008 Geochem.Trans.9 4
[8]Kerist S,Felmy A R,Ilton E S 2011 Environmental Science&Technology 45 2770
[9]Krese G,Furthmuller J1996 Phys.Rev.B 54 11169
[10]Kresse G,Joubert D 1999 Phys.Rev.B 59 1758
[11]Kresse G,Hafner J1994 Phys.Rev.B 49 14251
[12]Kresse G,Hafner J1993 Phys.Rev.B 47 RC558
[13]Blochl PE 1994 Phys.Rev.B 50 17953
[14]Ziesche P,Eschrig H 1991 Electronic Structure of Solids(Berlin:Akademie)p6
[15]Perdew JP,Chevary JA,Vosko SH,Jackson,Pederson M R,Singh D J,Fiolhais C 1992 Phys.Rev.B 46 6671
[16]Baer Y,Schoenes J1980 Solid State Commun.33 885
[17]Dudarev SL,Botton GA,Savrasov SY,Szotek Z,Temmerman WM,Sutton A P1998 Phys.Status Solidi A 166 429
[18]Anisimov V I,Zaanen J,Andersen OK 1991 Phys.Rev.B 44 943
[19]Liechtenstein A I,Anisimov V I,Zaanen J1995 Phys.Rev.B 52 5467
[20]Monkhorst H J,Pack JD 1977 Phys.Rev.B 16 1748
[21]Domain C,Bessonb R,Legris B 2002 Acta Materialia 50 3513
[22]Cai J,Lu D G 2013 Acta Metall.Sin.(Engl.Lett.)26 25
猜你喜欢
大学物理(2022年9期)2022-09-28
教学考试(高考化学)(2022年4期)2022-08-30
椰城(2021年12期)2021-12-10
陶瓷学报(2021年3期)2021-07-22
少儿科学周刊·少年版(2021年22期)2021-01-17
物理通报(2020年7期)2020-07-01
新世纪智能(数学备考)(2019年9期)2019-10-16
物理学报(2017年21期)2017-11-10
物理学进展(2017年1期)2017-02-23
腐蚀与防护(2016年7期)2016-09-14
