
人工锌指核酸酶突变EGFP基因的功能分析
2013-11-12 02:20袁玉国于宝利宋绍征周峰张利清顾迎迎禹明慧成勇
生物工程学报 2013年11期
袁玉国,于宝利,宋绍征,周峰,张利清,顾迎迎,禹明慧,成勇
扬州大学兽医学院 江苏省动物重要疫病与人兽共患病防控协同创新中心,江苏 扬州 225009
基因组靶向修饰是近年来生命科学研究的热点之一,特别是在基因功能分析、生产模式动物、治疗人类疾病和优良家畜育种等方面,基因组靶向修饰具有广阔的应用前景[1]。应用同源重组可实现对目的基因靶向修饰,但由于其费时、效率低 (10-5~10-7)等缺点,成为限制该项技术广泛应用的瓶颈之一[1-2]。利用人工锌指核酸酶(Zinc-finger nucleases,ZFNs)可引入DNA定点DSBs (Double-strand breaks,DSBs)以高效介导基因定点突变,极大提高基因打靶效率 (甚至可达到 10%以上效率),从而给基因组靶向修饰带来了新的希望[2-4]。ZFNs由一个识别特定DNA序列的锌指蛋白域和非特异性核酸内切酶 (FokⅠ)组成,其作用机理是 DNA结合域与特定 DNA序列结合,与之相连的非特异性核酸内切酶随之发挥剪切作用,在结合位点产生断裂,促进同源重组,提高定点突变和置换频率[5]。每个锌指蛋白可特异性识别并结合DNA链上的3个连续碱基,一个由3个及以上的锌指蛋白组成的锌指蛋白域可结合9个及以上碱基长度的靶位点,并且改变锌指蛋白中的几个残基可产生新的具有不同DNA结合特异性的ZFNs[6]。因此,可通过基因工程改造锌指结构域使锌指核酸酶针对复杂基因组里的特定DNA序列,借助内源DNA的修复机制可精确修饰高等生物的基因组,不仅可以将基因组靶向修饰的效率提高几个数量级,而且具有极高的特异性[7-8]。随着ZFNs介导基因打靶技术的发展和成熟,农业育种和包括为人类疾病和器官移植的大动物模型在内的医学研究等将毫无疑问受益于此项新技术[1,4]。在基因和细胞治疗方面,利用ZFNs对人体细胞、胚胎干细胞和诱导的多能干细胞的不同靶基因座进行靶向修饰,均获得了较好的效率[9-10]。
目前,哺乳动物转基因大多采用体细胞克隆的方法,在此过程中,筛选转基因细胞必须应用抗性或标记基因,这就使得获得的转基因动物携带抗性或标记基因,但转基因动物抗性或标记基因的生物安全性是一个必须解决的问题,而探索应用ZFNs进行标记基因删除的研究是一项极有意义的工作。目前,ZFNs是否能介导山羊体细胞基因组靶向修饰以及修饰方式未见报道,本实验构建并表达针对EGFP基因的ZFNs进行转基因山羊胎儿成纤维细胞打靶的研究,以揭示ZFNs靶向修饰 EGFP基因的方式和效率,为ZFNs结合体细胞核移植技术进行转基因动物标记基因敲除、提高生物安全性提供新思路。
1 材料与方法
1.1 材料
质粒pMLM290 (Addgene plasmid 21872)和pMLM292 (Addgene plasmid 21873)购自addgene公司;质粒 pAPLM由本实验室保存;实验羊购自高邮市菱唐乡;各种限制性内切酶、DNA连接试剂盒购自大连宝生物工程有限公司;DNA纯化试剂盒购自 QIAGEN公司;转染液购自 Eppendorf公司;细胞培养相关试剂购自HyClone公司;核移植相关试剂购自 Sigma公司。
1.2 人工锌指核酸酶真核表达载体的构建
将 pMONO-neo-GFP基因序列 (www.invivogen.com)输入到 CoDA软件 (www.zifit.partners.org)中进行分析,选取EGFP编码序列上的一个靶位点 (ACAATCTTCtttaagGATGA TGGA)进行锌指蛋白表达载体的构建 (表 1)。人工合成表达识别靶序列左右侧3个三联体氨基酸序列的DNA (EGFP-L ZFNs sequence,EGFP-R ZFNs sequence,见图1),用XbaⅠ和BamHⅠ双酶切后,凝胶回收276 bp片段。同时用XbaⅠ和BamHⅠ分别双酶切 pMLM290和 pMLM292质粒,线性化长片段再用连接酶与上述的 276 bp片段分别连接构建成左右侧载体。构建正确的载体用 ApaLⅠ和 NruⅠ双酶切,胶回收 3 313 bp片段用于细胞打靶的研究。
1.3 转基因克隆胎儿成纤维细胞系的建立
用SalⅠ和NotⅠ酶切、胶回收含EGFP表达框的质粒pAPLM片段[11],稀释成2~3 ng/µL用于单细胞基因注射。在Eppendorf显微操作系统中进行胎儿成纤维细胞基因注射,具体方法见文献[12]。G418筛选后获得的转基因细胞作供核细胞。山羊超数排卵、手术取卵、卵母细胞去核、移核及胚胎移植操作方法同文献[13]。移植受体35 d B超妊娠检查,怀孕受体手术取胎儿建立转基因克隆胎儿成纤维细胞系 (2#),方法同前。细胞冻存于液氮中备用。
1.4 ZFNs电转染转基因胎儿成纤维细胞及FACS分析
复苏2#细胞,培养至70%~80%汇合时进行电转染含左右侧3 313 bp的ZFNs,条件同上。同时共转染用ApaLⅠ和NruⅠ双酶切pMLM290和pMLM292片段作对照。转染细胞电转染后分2份,一份稀释成单细胞培养在96孔板中,另一部分培养在六孔板中,在第6天时,0.25%胰酶消化细胞,加D-hank’s吹打分散、离心,重复3次,用含1% FCS的 D-hank’s吹打细胞,然后细胞悬浮于D-hank’s中,以1×106的细胞数进行FACS荧光分析,实验重复3次。
1.5 细胞EGFP基因序列突变分析
96孔板中细胞长满后,GFP观察呈阴性的细胞传至48孔板,生长至90%汇合时,收集细胞,用天根细胞基因组 DNA提取试剂盒提取DNA。以DNA为模板,用跨突变区的特异性引物 (Forward: 5¢-GCTGGATGGTGATGTGAATG G-3¢; Reverse: 5¢-GTGTTCTGCTGGTAATGGTC TGC-3¢)PCR扩增484 bp产物,扩增条件为:94 ℃ 5 min ;94 ℃ 30 s,62 ℃ 40 s,72 ℃ 40 s;72 ℃延伸5 min。然后将PCR产物克隆至T载体上,挑取10个克隆送上海生工生物公司测序。

表1 EGFP上ZFNs靶位点及其锌指氨基酸序列Table 1 EGFP ZFNs target sites and amino-acid sequences of designed ZF recognition helices

图1 ZFNs识别区左右侧编码序列Fig. 1 Left and right coding sequence of ZFNs.
2 结果
2.1 人工锌指核酸酶真核表达载体的构建
将EGFP基因序列输入CoDA软件中,将条件设置5~6个,进行分析,找到一个锌指蛋白识别编码绿色荧光蛋白基因的靶位点,如表2所示左侧3个三联体DNA是TGTGATGAA,右侧3个三联体DNA是GGAGATGAT,左右侧3个锌指蛋白所对应的氨基酸序列和编码氨基酸所对应的DNA序列见EGFP-R ZFNs和EGFP-L ZFNs sequence,斜体序列为酶切位点。人工合成这两段 DNA序列,用 XbaⅠ和BamHⅠ双酶回收后分别插入到pMLM290和pMLM292载体中,构建成一对锌指核酸酶表达载体 pLZFE1729和pRZFE1729 (图2),用BamHⅠ酶切均可有5 973 bp条带 (图3),与预期结果相符。
2.2 转基因克隆胎儿成纤维细胞系的建立
运用单细胞基因显微注射的方法对培养的贴壁细胞进行注射,细胞经G418筛选后获得了两个单克隆细胞株,且在荧光显微镜下均可观察到绿色荧光。用这两株细胞进行了2次核移植实验,共超排了8只供体山羊,获得了57枚MII期卵母细胞,将29枚激活胚移植到4只受体中,其中3只35 dB超检查呈阳性,取2#受体羊胎儿,建立了2#-1和2#-2两个胎儿细胞系。
2.3 打靶细胞FACS分析

图2 pLZFE1729和pRZFE1729质粒图Fig. 2 Plasmids of pLZFE1729 and pRZFE1729. CMV: CMV promoter; NLS: SV40 nuclease localization signal; ZF:sequence of EGFP-L ZFNs sequence and EGFP-R ZFNs; Fok I: Fok I nuclease; PA: BGH polyadenylation sequence;SV40: SV40 early promoter and origin; Blas: blasticidin resistance gene.
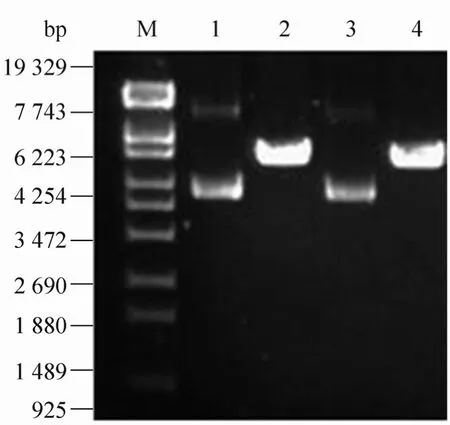
图3 质粒pLZFE1729和pRZFE1729的酶切鉴定Fig. 3 Identification of pLZFE1729 and pRZFE1729 by enzyme digestion. M: marker T14; 1, 3: plasmid of pLZFE1729 and pRZFE1729; 2, 4: pLZFE1729 and pRZFE1729 digested with BamH I.
pLZFE1729和pRZFE1729载体酶切回收后共电转染2#-1细胞,在第6天收集细胞进行FACS分析,转染锌指核酸酶的细胞仅有 0.2%细胞有绿色荧光,而对照组细胞中有5%的细胞有可见绿色荧光 (图4),两者存在差异,表明构建的锌指核酸酶转染后对细胞EGFP基因有切割左右或突变作用,导致细胞发绿色荧光的比例减少。
2.4 突变靶序列测序分析
PCR扩增细胞转染后的靶序列区域,产物测序结果与正常序列经Blast软件比对结果显示,在外显子GTTAC (1 894 bp位置)后增加一个碱基G (图5),将突变后的EGFP编码序列输入蛋白翻译软件进行分析,编码框发生改变,其相应的氨基酸由原先的239个变成156个。
3 讨论
目前构建人工锌指核酸酶的方法有模块组装 (Modular assembly,MA)、寡聚文库构建(Oligomerized pool engineering,OPEN)、上下文依赖组装 (Context-dependent assembly,CoDA)等方案[14],为方便广大研究人员,锌指协会开发了一个锌指靶序列搜索工具 (Zinc finger targeter,ZiFiT,http://zifit. partners.org/ZiFiT/),输入特定序列并设置一定参数,可用于寻找到适宜于这3种方案的的3-三联子目标序列,并对所得靶位点构建 ZFN成功可能性进行评估。本实验运用CoDA方法将EGFP编码序列输入到该网站上进行分析,在编码序列上找到一个靶位点,并将获得的三联子对应 DNA序列连接到含有FokⅠ核酸内切酶的通用质粒 (pMLM290和pMLM292)中,即构建成一对人工锌指核酸酶表达质粒 (pLZFE1729和pRZFE1729)。

图4 FACS分析绿色荧光强度Fig. 4 Analysis for EGFP fluorescence intensity by FACS. In the ZFNs-treated cells (S1-1796, S1-1796-2), the number of cells expressing of EGFP was 0.2%. In the control (S1-1796-3), the number of cells expressing of EGFP was 5%.

图5 突变细胞PCR产物序列Blast比对结果Fig. 5 Blast result of PCR product of mutant cells.
基因敲除或突变以研究基因功能和获得动物模型是一项非常有用的方法。传统的基因敲除技术如同源重组效率低且需要长期的药物筛选,目前仅在小鼠和人上获得胚胎干细胞,因而限制了该技术在家畜上的应用。培养体细胞打靶效率低也是体细胞核移植技术较难应用于大型家畜上的主要原因。随着ZFNs技术的发展和逐渐成熟,其在哺乳动物基因打靶上的优势已逐渐显现。通过显微注射的方法将ZFNs质粒或mRNA注射到胚胎原核,出生大鼠基因敲除效率最高可达 40%,并且可通过生殖系遗传给后代[15]。由于用显微注射方法产生的动物存在嵌合体等缺点,且需要大量的受体、繁殖周期长,并需要大量的饲养成本,对于大型哺乳类动物而言,该方法存在很大的风险[16]。针对细胞中特定基因设计特异性 ZFNs,通过对细胞进行电转染 ZFNs质粒或 mRNA可提高打靶效率,同时结合体细胞核移植技术获得基因敲除动物[16-19],可解决传统的同源重组技术由于长期药物筛选导致细胞活力差和获得更多的单克隆打靶细胞系等问题。为了验证所构建的ZFNs有效性,同时为以后获得不含荧光蛋白山羊奠定基础,本实验将ZFNs片段转染含EGFP基因的原代克隆山羊胎儿成纤维细胞,与未转染组相比,细胞发绿色荧光的比例降低了4.8%,初步证明了构建的ZFNs有效性。Watanable等[20]用转基因猪原代胎儿成纤维细胞的EGFP基因进行ZFNs打靶,其细胞发绿色荧光的比例降低了15%,与本实验获得的结果基本相似,应用ZFNs消除或降低荧光蛋白的模式不仅在猪上有效,而且在山羊上也可得到类似的结果。作者推测获得的转基因克隆胎儿成纤维细胞发荧光的比例较低,可能与基因的甲基化或乙酰化而降低了对照细胞的荧光数目所导致,与我们前期的报道类似[13]。
ZFNs技术是近几年发展起来的转基因技术,其对基因靶向敲除的效率和特异性有待进一步提高,尤其对靶位点的突变存在如碱基缺失、插入等多种方式,甚至存在脱靶现象也时常发生。本实验对打靶细胞PCR产物测序结果表明,靶序列及附近上下游序列无突变,仅在距靶序列下游180 bp左右位置插入1个碱基G,这种突变方式可能由于构建的ZFNs特异性较差而存在脱靶现象。Watanable等报道其 EGFP序列突变方式有碱基插入、缺失和替换,并且突变发生在靶序列位置或其上、下游附近位置,其中插入的碱基为4 bp,缺失的碱基为1~15 bp,替换的碱基为1 bp。同样在猪、牛等哺乳动物细胞内源基因上也同样存在类似的结果[16-19]。在本实验中,构建的ZFNs产生的突变方式虽然在距靶序列较远位置插入1个碱基G,但其突变仍发生在编码绿色荧光蛋白的外显子位置上,不仅造成 83个氨基酸缺失,而且从152~156的5个氨基酸发生突变,其表达的蛋白分子量大小由正常的26.9 kDa变成17.6 kDa,这可能是导致有荧光细胞减少的分子生物学原因。从实验结果来看,ZFNs转染细胞靶位点虽然出现异位现象,但在特异性位点附近还是发生了突变,并使绿色荧光细胞比例下降,什么原因导致脱靶需要进一步研究。
[1]Provost FL, Lillico S, Passet B, et al. Zinc fi nger nuclease technology heralds a new era in mammalian transgenesis. Trends Biotechnol, 2009,28(3): 134-140.
[2]Meyer M, de Angelis MH, Wurst W, et al. Gene targeting by homologous recombination in mouse zygotes mediated by zinc-finger nucleases. Proc Natl Acad Sci USA, 2010, 107(34): 15022-15026.
[3]Urnov FD, Rebar EJ, Holmes MC, et al. Genome editing with engineered zinc finger nucleases. Nat Rev Genet, 2010, 11(9): 636-646.
[4]Rémy S, Tesson L, Ménoret S. Zinc-finger nucleases: a powerful tool for genetic engineering of animals. Transgenic Res, 2010, 19(4): 363-371.
[5]Liu PQ, Chan EM, Cost GJ, et al. Generation of a triple-gene knockout mammalian cell line using engineered zinc-f i nger nucleases. Biotechnol Bioeng, 2010, 106(6): 97-105.
[6]Beumer KJ, Trautman JK, Bozas A, et al. Eff i cient gene targeting in Drosophila by direct embryo injection with zinc-f i nger nucleases. Proc Natl Acad Sci USA, 2008, 105(12): 19821-19826.
[7]Miller JC, Holmes MC, Wang J, et al. An improved zinc-finger nucle ase arch itecture f or highly specic genome editing. Nat Biotechnol, 2007, 25(4): 778-785.
[8]Doyon Y, McCammon JM, Miller JC, et al.Heritable targeted gene disruption in zebrash using designed zinc-finger nucleases. Nat Biotechnol,2008, 26(6): 702-708.
[9]Hockemeyer D, Soldner F, Beard C, et al. Eff i cient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-f i nger nucleases. Nat Biotechnol, 2009, 27(5): 851-857.
[10]Zou J, Maeder ML, Mali P, et al. Gene targeting of a disease-related gene in human induced pluripotent stem and embryonic stem cells. Cell Stem Cell,2009, 5: 97-110.
[11]Yuan YG, An LY, Yu BL, et al. Construction of mammary gland specific vector for expression of human lactoferrin. J Yangzhou Univ: Agri Life Sci Ed, 2011, 32(3): 6-10 (in Chinese).袁玉国, 安礼友, 于宝利, 等. 人乳铁蛋白乳腺特异性表达载体的构建及其功能的验证. 扬州大学学报: 农业与生命科学版, 2011, 32(3): 6-10.
[12]Gueroussov S, Tarnawsky SP, Cui XA, et al.Analysis of mRNA nuclear export kinetics in mammalian cells by microinjection. J Vis Exp,2010, 1(46): 2387-2395.
[13]An LY, Yuan YG, Yu BL, et al. Generation of human lactoferrin transgenic cloned goats using donor cells with dual markers and a modified selection procedure. Theriogenology, 2012, 78(6):1303-1311.
[14]Xiao A, Hu YY, Wang WY, et al. Progress in zinc finger nuclease engineering for targeted genome modification. Hereditas, 2011, 33(7): 665-683 (in Chinese).肖安, 胡莹莹, 王唯晔, 等. 人工锌指核酸酶介导的基因组定点修饰技术. 遗传, 2011, 33(7):665-683.
[15]Geurts AM, Cost GJ, Freyvert Y, et al. Knockout rats via embryo microinjection of zinc-finger nucleases. Science, 2009, 325(5939): 433.
[16]Yang D, Yang H, Li W, et al. Generation of PPARγ mono-allelic knockout pigs via zinc-finger nucleases and nuclear transfer cloning. Cell Res,2011, 21(6): 979-982.
[17]Hauschild J, Petersen B, Santiago Y, et al. Efficient generation of a biallelic knockout in pigs using zinc-finger nucleases. Proc Natl Acad Sci USA,2011, 19, 108(29): 12013-12017.
[18]Whyte JJ, Zhao J, Wells KD, et al. Gene targeting with zinc finger nucleases to produce cloned eGFP knockout pigs. Mol Reprod Dev, 2011, 78(1): 2.
[19]Yu S, Luo J, Song Z, et al. Highly efficient modification of beta-lactoglobulin (BLG)gene via zinc-finger nucleases in cattle. Cell Res, 2011,21(11): 1638-1640.
[20]Watanabe M, Umeyama K, Matsunari H, et al.Knockout of exogenous EGFP gene in porcine somatic cells using zinc-finger nucleases. Biochem Biophys Res Commun, 2011, 402(1): 14-18.
猜你喜欢
学与玩(2022年10期)2022-11-23
今日农业(2022年3期)2022-06-05
昆明医科大学学报(2022年1期)2022-02-28
实用临床医药杂志(2021年13期)2021-01-10
无机化学学报(2020年7期)2020-07-20
中国预防兽医学报(2020年2期)2020-06-01
三农资讯半月报(2020年8期)2020-05-13
中国病理生理杂志(2017年2期)2017-01-17
创新科技(2015年1期)2015-12-24
中华胰腺病杂志(2015年5期)2015-12-08
