
铁催化芳基格氏试剂的联芳交叉偶联的反应机理
2015-02-18 12:06任清华沈晓燕
物理化学学报 2015年5期
任清华 沈晓燕
(上海大学理学院化学系,上海200444)
1 lntroduction
Transition metal-catalyzed cross-coupling reactions are of utmost importance for the formation of carbon-carbon as well as carbon-heteroatom bonds.In general,palladium and nickel catalysts dominate this field,such as,Negishi,1,2Suzuki,3,4Heck,5,6Stille,7,8and Kumada-Corriu9-11reactions.The biaryl compounds are very important intermediates in modern organic synthesis and pharmaceutical products.12Palladium-and nickel-catalyzed crosscoupling reactions are among the most versatile methods for the synthesis of biaryl compounds.Suzuki-Miyaura reactions between arylboronic acids and aryl halides are widely applied because of their especially mild conditions and excellent compatibility with many functional groups.13-16However,palladium and nickel catalysts are limited by the high price or considerable toxicity,especially for industrial processes.Iron-catalyzed cross-coupling reactions have become increasingly important in the construction of complex molecular frameworks in the last few decades due to their high efficiency,cheapness,non-toxicity,and the environmental health of the simple iron salt precatalysts.17-20The complex,airsensitive ligands are also not required and the mild reaction conditions tolerate various functional groups.
Furstneret al.21,22reported a lot of highly selective iron-catalyzed cross-coupling reactions of aryl halides with alkyl magnesium halides and the iron-catalyzed cross-coupling reactions have matured into a class of effective carbon-carbon as well as carbon-heteroatom bond-forming reactions.23-27In general,the iron-catalyzed biaryl syntheses from aryl Grignard species and aryl halides are proved unselective.22,28-30For example,Hatakeyamaet al.31,32reported a method which requires a special precatalyst combination of iron(II)fluoride and a carbine ligand and also needs the generation of the catalytically active species in a prior step to improve the selectivity.
Later,von Wangelinet al.33paid attention to many papers34,35about the rich coordination abilities of olefins with iron in low oxidation states and proposed a new method of activation for olefin-substituted aryl chlorides.The sequential olefin coordination at the catalyst and haptotropic migration could affect C―Cl bond activation of chlorostyrenes.36They had reported an efficient iron-catalyzed biaryl synthesis where the reaction of aryl Grignard species with chlorostyrenes proceeded under mild reaction conditions in the presence of iron(III)tris(acetylacetonate)[Fe(acac)3]as precatalyst.36
The theoretical understanding of the reaction mechanisms of the transition metal-catalyzed processes may shed light on how to design more powerful catalysts and how to control the catalysis process.However,the mechanisms of the iron-catalyzed reactions remain rather obscure.Kochi,37Furstner,20,22Bogdanovic,38Norrby,39et al.have reported some mechanisms of iron-catalyzed crosscoupling reactions of Grignard reagents.Our group40have investigated the mechanisms of iron-catalyzed cross-coupling reactions of alkyl Grignard reagents using density functional theory calculations.
The catalyzed cycles start form the inorganic Grignard reagent[Fe(MgX)2],where X=Br or I.[Fe(MgX)2]consists of two magnesium atoms and one iron center that are connectedviatwo covalent intermetallic bonds.It was generally used to explain many of the experimental results by Furstneret al,20-22where the initial[Fe(MgX)2]is formed from the iron precatalyst and RMgX.The mechanism is through Fe(-II)and Fe(0)catalytic cycle,where the iron center is Fe(-II)in[Fe(MgX)2],and Fe(0)in[1RFe(MgX)]and[1R-(2R)-Fe(MgX)2].The multiplicity of Fe is set as 1.As far as we know,detailed theoretical studies concerning the[Fe(MgX)2]catalyzed biaryl cross coupling processes of aryl Grignard reagents have not yet been performed,although the experimental results involving iron-catalyzed cross-coupling of aryl Grignard reagents have recently been reported.41,42
Here,we select one typical reaction in the experimental work of Gulak and von Wangelin36as an example,which is depicted in Scheme 1,in order to gain insight into the mechanisms of ironcatalyzed cross-coupling reactions of aryl Grignard reagents.The experimental reaction was carried out in tetrahydrofuran(THF)/N-methyl-2-pyrrolidone(NMP)using Fe(acac)3(5%,molar fraction)as the precatalyst.Fe(acac)3reacts with R-MgBr to yield the inorganic Grignard reagent[Fe(MgBr)2],which is readily soluble in THF.Our thorough theoretical investigation is applied to[Fe(MgBr)2]catalyzed cross-coupling reaction betweenorthochlorostyrene[Cl-Ph-vinyl]and phenylmagnesium bromide[PhMgBr]using density functional theory(DFT)calculations with the B3LYP method.43-48
We will study two mechanisms.One mechanism is named as Cycle A with forming[Ar-Fe(MgBr)]in the catalytic cycle and another mechanism is named as Cycle B without forming[Ar-Fe(MgBr)]in the catalytic cycle.Our theoretical studies will answer the questions:Which catalytic cycle is favored?Which step is the rate-determining step in the whole catalytic cycle?Is it possible to lower the energy barrier when considering the solvent effect?Answers to these questions will improve the understanding of ironcatalyzed cross-coupling of aryl Grignard reagents.
2 Computational methods
All DFT calculations were performed using the Gaussian 03 program49with the B3LYPhybrid functional.43-48The 6-311++G(d,p)basis set was used for C,H,Cl,Br,and Mg.50,51The SDD quasirelativistic pseudopotential and associated basis set was used for iron.52All equilibrium structures of the gas phase geometries were fully optimized without any symmetry restriction and then the vibrational frequencies analysis was performed to ensure that the local minima had zero imaginary frequencies and the transition state had exactly one.The solvent effect was calculated using the conductor polarized continuum model(CPCM)method53with the UFF radii on the gas-phase optimized geometries.The polarizable continuum model(PCM)is a commonly used method in computational chemistry to model salvation effects.It models the solvent as a polarizable continuum,rather than individual molecules,which has two popular types:dielectric PCM(DPCM)and conductor-like PCM(CPCM).Both the electronic and nonelectronic free energies in solution were added to the gas-phase Gibbs free energies to obtain the solution Gibbs free energies in THF,ΔGsol.40All the solution-phase free energies reported in the paper correspond to the reference state of 1 mol∙L-1,298 K.

Scheme 1 Iron-catalyzed cross-coupling reaction between ortho-chlorostyrene and phenylmagnesium bromide
3 Results and discussion
3.1 Catalytic Cycle A with forming[Ar-Fe(MgBr)]
The catalytic cycle A is similar as the mechanistic proposal of Furstneret al.,22which is outlined in Fig.1.It includes three basic steps:(I)oxidation of[Fe(MgBr)2]to obtain[Ar-Fe(MgBr)],(II)addition with phenylmagnesium bromide to yield[Ar-(phenyl)-Fe(MgBr)2],and(III)reductive elimination to return to[Fe(MgBr)2].The cycle A includes three transition states(TS1,TS2,TS3).The fully optimized structures of the reactant R1,the catalyst CA,the complex 2,the transition state TS1,the intermediates 3 and 4,and the product P2are shown in Fig.2.The fully optimized geometries of the reactant R2,the complex 5,the transition states TS2,TS3,the intermediate 6,and the final product P1are shown in Fig.3.The values of the bonding distances(the unit is nm)are also given in Figs.2 and 3.
(I)oxidation of[Fe(MgBr)2]to obtain[Ar-Fe(MgBr)].The approach of R1toward the catalyst CA,[Fe(MgBr)2],leads to the formation of a complex 2.From 2,after passing through a transition state TS1,the intermediate 3 of oxidative addition is obtained.In the optimized geometry of 2(see Fig.2),the iron atom is above the center of the phenyl and is far from the1C―Cl bond.The distances of1C―Fe and Cl―Fe are 0.207 and 0.342 nm,respectively.Then in the transition state TS1,the chlorine atom approaches toward2Mg atom around the iron center departing from1C atom.The distance of Cl―Fe is 0.239 nm.It becomes 0.224 nm in the optimized geometry of 3,accompanying with the formation of Cl―2Mg bond(0.242 nm,see Fig.2).At the same time,the distance of Fe―2Mg increases from 0.253 nm in the optimized structure of 2 to 0.260 nm in the optimized structure of 3 because of the bonding of Cl atom with the2Mg atom and the interaction of Cl atom with Fe atom.The weakening process of the bonding of Fe―2Mg is beneficial for the further reaction,i.e.,the covalent intermetallic bonding of Fe―2Mg breaks and the molecule P2dissociates from 3 to form the intermediate 4,[(BrMg)-Fe-(Ph-vinyl)].The bond distances of1C―Fe and Fe―1Mg in the optimized structure of 4 are 0.189 and 0.245 nm,respectively.
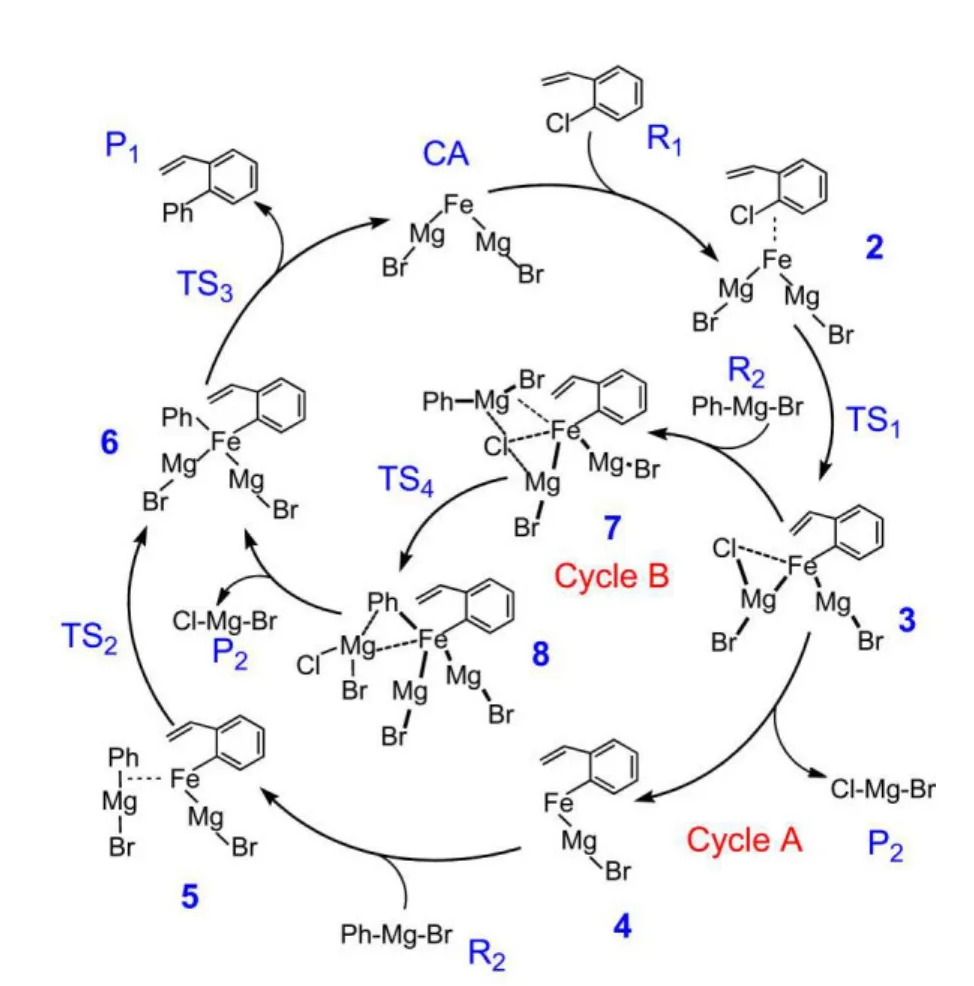
Fig.1 Outline of the iron-catalyzed cross-coupling reaction mechanism for the product P1from ortho-chlorostyrene R1and phenylmagnesium bromide R2

Fig.2 Fully optimized structures of the reactant R1,the catalyst CA,the intermediates 2,3,4,the transition state TS1,and the product P2
(II)Addition of another reactant phenylmagnesium bromide R2to 4 yields the intermediate 6,[Ar-(Ph-)-Fe(MgBr)2].After the intermediate 4 is formed,the approach of R2toward 4,leads to the formation of a complex 5.From 5,passing through a threemembered-ring transition state TS2,the intermediate 6 is formed.From the fully optimized structures of 5,TS2and 6 shown in Fig.3,it can be seen that the Fe―2Mg distance is 0.267 nm in TS2,compared to that of 0.277 nm in 5.The distance of Fe―2C also decreases from 0.439 nm in 5 to 0.354 nm in TS2,so that it is easy for atom2C to attack Fe atom in TS2,leading to the formation of a Fe―2C bond to obtain the intemediate 6.The bond distance of Fe―2C in 6 is 0.189 nm.
(III)Finally,reductive elimination of 6 to return to the catalyst CA,[Fe(MgBr)2].This process passes through the third transition state TS3to form the product P1,[Ph-Ph-vinyl]and to regenerate the original catalyst CA.From the fully optimized structures of 6,TS3,and P1shown in Fig.3,the2C and1C atoms in TS3go closer to 0.179 nm from 0.287 nm in 6 and finally the bond of2C―1C is formed in P1(0.149 nm).
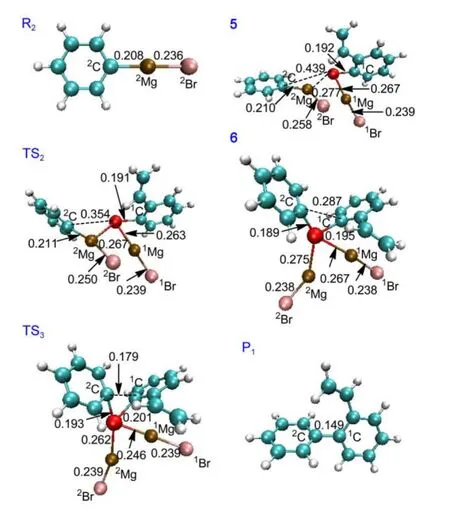
Fig.3 Fully optimized structures of the reactant R2,the intermediates 5,6,the transition states TS2,TS3,and the final product P1
The electronic energy profiles for the whole catalytic cycles for iron-catalyzed cross-coupling reaction betweenortho-chlorostyrene and phenylmagnesium bromide are depicted in Fig.4,where the summation of the electronic energies of the reactant R1and the catalyst CAis set as zero energy.
From Fig.4,it can be seen that the energy barrier from the complex 2 to the first transition state TS1is 106.11 kJ∙mol-1.The energy value needed for the process in which the molecule P2,[Cl-Mg-Br],dissociates from the intermediate 3 to form the intermediate 4 is 115.16 kJ∙mol-1.It refers to the breaking of the strong covalent intermetallic bonding of Mg―Fe.This could be offset by the large exothermicity(-139.64 kJ∙mol-1)process forming the intermediate 3 from 2.The transition state connecting 3 and 4 has not been found after many attempts.The energy barrier of TS2from 5 to form a stable structure 6[Ar-(phenyl)-Fe-(MgBr)2]is small(4.87 kJ∙mol-1).
The energy barrier of TS3to form the final product P1from the intermediate 6 when releasing the catalyst CA through the reductive elimination is 96.47 kJ∙mol-1.This process is needed to break two Fe―C bonds.In all,the energy barriers for the steps of 2➝3,3➝4 and 6➝CA+P1are closer and the step of 3➝4 is the rate-limiting step in the whole catalytic cycle of the mechanism of CycleAfor the gas calculation.
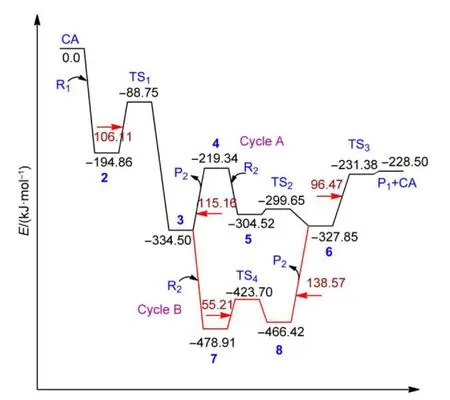
Fig.4 Electronic energy profiles for the overall catalytic cycles of the iron-catalyzed cross-coupling reaction
3.2 Catalytic cycle B without forming[Ar-Fe(MgBr)]
Considering that the required electronic energy for the process in which the molecule P2,[Cl-Mg-Br]dissociates from the intermediate 3 to form the intermediate 4 is very high by 115.16 kJ∙mol-1(see Fig.4),it could be possible that there is another way to lower the energy barrier,namely if phenylmagnesium bromide(R2)attacks the intermediate 3 directly before the formation of 4,[Ar-Fe(MgBr)].Our previous calculations40about[Fe(MgBr)2]catalyzed cross-coupling reaction between 4-chlorobenzoic acid methyl ester andn-hexylicmagnesium bromide have proved that the new path is easier than the catalytic cycleAwith forming[Ar-Fe(MgBr)].We did the similar calculations for the reaction betweenortho-chlorostyrene and phenylmagnesium bromide.The catalytic cycle is named as cycle B,which is also shown in Fig.1.
From Fig.1,it can be seen that the reactant R2firstly approaches toward the intermediate 3 to form the complex 7.From 7,after passing through one transition state TS4,an intermediate 8 is formed.The fully optimized structures of 7,TS4and 8 in the gas phase are shown in Fig.5.Then the molecule P2dissociates from the intermediate 8 to yield 6.In the optimized geometry of 7,the distance of Cl―3Mg is 0.245 nm.The Cl―2Mg bond length becomes longer to 0.264 nm compared to that of 0.242 nm in 3,which means that the Cl atom is now linked to the3Mg atom.The distance of Cl with Fe also becomes longer to 0.241 nm,compared to that of 0.224 nm in 3.The distances of Fe―3Br and Fe―3Mg are 0.250,0.345 nm,respectively.Next,the atom3Br in 7 rotates around the3Mg atom to bring the atom2C close to the Fe atom,so that the atom2C can attack the Fe center easily.In the optimized structure of the transition state TS4,the distance of atom2C from the Fe atom is 0.258 nm.It becomes shorter to 0.201 nm in the optimized structure of 8,which is close to the value of 0.189 nm in the optimized structure of 6(see Fig.3).The distances of2C―3Mg and Fe―3Mg become longer to 0.225,0.316 nm in 8,compared to those of 0.210,0.264 nm in TS4,respectively.It is beneficial for the process in which the molecule P2dissociates from the intermediate 8 to yield 6.

Fig.5 Fully optimized structures of the intermediates 7,8 and transition state TS4for the Cycle B
The electronic energy profile from the intermediate 3 to the intermediate 6 for Cycle B is also depicted in Fig.4.From Fig.4,it can be seen that the energy barrier to pass over the transitional state TS4is only 55.21 kJ∙mol-1,which is much smaller than 115.16 kJ∙mol-1required when the molecule P2,[Cl-Mg-Br]dissociates directly from the intermediate 3 before another reactant R2attacks.However,the energy needed for the process when the molecule P2,[Cl-Mg-Br]dissociates from the intermediate 8 to form 6 is very high,i.e.,138.57 kJ∙mol-1.It looks that the Cycle B is not favored than CycleAfor the gas phase calculations,which is different from the previous calculations about[Fe(MgBr)2]catalyzed cross-coupling reaction betweenn-hexylicmagnesium bromide and 4-chlorobenzoic acid methyl ester.40
3.3 Solvent effect
All of the above results are calculated in the gas phase.According to our previous study about[Fe(MgBr)2]catalyzed crosscoupling reaction between 4-chlorobenzoic acid methyl ester andn-hexylicmagnesium bromide,40the CPCM method is suitable to calculate the solvent effect for the iron-catalyzed cross-coupling reactions of alkyl Grignard reagents.Now we used the same CPCM method to investigate the solvent effect for the[Fe(MgBr)2]catalyzed cross-coupling reaction betweenortho-chlorostyrene and phenylmagnesium bromide.In order to clarify whether the solvent effect can play a significant role in the process,we compare the Gibbs free energies ΔGgand the Gibbs free energies in THF solution,ΔGsolfor the overall catalytic cycles of two mechanisms(Cycle A and Cycle B),which are shown in Fig.6.The former is depicted as the solid line and the latter is drawn as the dashed line.
From Fig.6,it can be seen that the solvent effect is very obvious for the steps of dissociating P2from 3 to form 4 in the CycleAor from 8 to obtain 6 in the Cycle B.ΔGsol(3➝4)is-55.94 kJ∙mol-1compared that ΔGg(3➝4)is 67.02 kJ∙mol-1,Similarly,ΔGsol(8➝6)is-17.00 kJ∙mol-1compared that ΔGg(8➝6)is 86.69 kJ∙mol-1.The solvent effect is exothermic.
The solvent effect causes large changes of the ΔGvalues for the processes of(3➝4)in the CycleAand of(8➝6)in the Cycle B.As far as we know,the solute is placed in a cavity in the solvent using CPCM method.The free energy of solvation was calculated from the surface charges of the solute and their interactions with the solvent.40,54In general,the solvent effect is large for the dissociation processes because the gas phase calculations strongly favor the formation of clusters and make dissociation reactions more difficult than in condensed phases.40,54-56For example,Schubert and Papai57has reported that the solvent effect was large for the process of dissociation of a PMe3ligand in the C―C coupling.Other calculations reported by Castellaniet al.58also showed that the solvent effect can change the dissociation energy value from 332.31 kJ∙mol-1in the gas phase to 168.45 kJ∙mol-1within the toluene solvent for the process of dissociation of μ-CH3contact ion-pair.

Fig.6 Gibbs free energy in gas phase(ΔGg,solid line)and Gibbs free energies in THF solution(ΔGsol,298 K,dashed line)for the overall catalytic cycles
We can see that the energy barrier ΔGsolfrom 4 through 5 to TS2in THF solvent during the process from 3 to 6 for CycleAis 39.76 kJ∙mol-1.However,the energy barrier ΔGsolfrom 7 through TS4to 8 in THF solvent during the process from 3 to 6 for Cycle B is only 7.74 kJ∙mol-1,which is smaller than that of CycleA.So the catalytic Cycle B without forming[Ar-Fe(MgBr)]is favored over the catalytic Cycle A with forming[Ar-Fe(MgBr)]when considering the solvent effect,this conclusion keeps line with our previous study.40
For the other steps from 2 to TS1and 6 to TS3,the solvent effect is also large,where the ΔGsol(2➝TS1)in THF is 68.90 kJ∙mol-1compared to ΔGg(2➝TS1)in the gas phase(96.98 kJ∙mol-1)and the ΔGsol(6➝TS3)in THF is 82.98 kJ∙mol-1compared to ΔGg(6➝TS3)in the gas phase(105.63 kJ∙mol-1).It can be seen that the solvent effects can lower the energy barrier.These variations can be clearly seen in Fig.6.
In all,the rate-limiting step in the whole catalytic cycles for both CycleAand Cycle B is the reductive elimination of 6 to form P1when the catalyst CA is re-generated,where the ΔGsolis 82.98 kJ∙mol-1in THF solvent using the CPCM method.The conclusion is the same as our previous study about[Fe(MgBr)2]catalyzed cross-coupling reaction between 4-chlorobenzoic acid methyl ester andn-hexylicmagnesium bromide.40The CPCM method has been widely used to study the solvent effect for transition metalcatalyzed cross-coupling reactions.59,60
4 Conclusions
Our studies reported the first theoretical investigation of[Fe(MgBr)2]catalyzed cross-coupling reaction betweenorthochlorostyrene and phenylmagnesium bromide using DFT calculations.We studied two mechanisms.One is named as Cycle A with forming[Ar-Fe(MgBr)]in the catalytic cycle.It includes three basic steps:(I)oxidation of[Fe(MgBr)2]to obtain the intermediate 4,[Ar-Fe(MgBr)],(II)addition to yield the intermediate 6,[Ar-(phenyl)-Fe(MgBr)2]and(III)reductive elimination to return to the catalyst CA,[Fe(MgBr)2].Another is named as Cycle B without forming[Ar-Fe(MgBr)]in the catalytic cycle.In the first step,the phenylmagnesium bromide R2attacks the intermediate 3 directly to form a new intermediate 7 before the molecule P2[Cl-Mg-Br]dissociates to form the intemediate 4,[Ar-Fe(MgBr)].
The solvent effects can lower the energy barrier.The catalytic Cycle B without forming[Ar-Fe(MgBr)]is favored over the catalytic Cycle A with forming[Ar-Fe(MgBr)]when considering the solvent effect.The rate-limiting step in the whole catalytic cycles for both Cycle A and Cycle B is the reductive elimination of 6 to form P1when the catalyst CA is re-generated,where the ΔGsolis 82.98 kJ∙mol-1in THF solvent using the CPCM method.
Supporting lnformation:Detailed molecule coordinates of all optimized structures and the calculated energy values are included.This information is available free of chargeviathe internet at http://www.whxb.pku.edu.cn.
(1) Negishi,E.Accounts Chem.Res.1982,15,340.doi:10.1021/ar00083a001
(2) Yang,Y.;Oldenhuis,N.J.;Buchwald,S.L.Angew.Chem.Int.Edit.2013,125,643.doi:10.1002/ange.201207750
(3) Blangetti,M.;Rosso,H.;Prandi,C.;Deagostino,A.;Venturello,P.Molecules2013,18,1188.doi:10.3390/molecules18011188
(4)Miyaura,N.;Suzuki,A.J.Chem.Soc.Chem.Commun.1979,19,866.doi:10.1039/C39790000866
(5) Heck,R.F.;Nolley,J.P.J.Org.Chem.1972,37,2320.doi:10.1021/jo00979a024
(6) Cabri,W.;Candiani,I.Accounts Chem.Res.1995,28,2.doi:10.1021/ar00049a001
(7) Milstein,D.;Stille,J.K.J.Am.Chem.Soc.1978,100,3636.doi:10.1021/ja00479a077
(8)Yabe,Y.;Maegawa,T.;Monguchi,Y.;Sajiki,H.Tetrahedron2010,66,8654.doi:10.1016/j.tet.2010.09.027
(9)Tamao,K.;Sumitani,K.;Kumada,M.J.Am.Chem.Soc.1972,94,4374.doi:10.1021/ja00767a075
(10)Yang,L.M.;Huang,L.F.;Luh,T.Y.Org.Lett.2004,6,1461.doi:10.1021/ol049686g
(11)Vechorkin,O.;Proust,V.;Hu,X.J.Am.Chem.Soc.2009,131,9756.doi:10.1021/ja9027378
(12) Torssell,K.B.Natural Product Chemistry:a Mechanistic and Biosynthetic Approach to Secondary Metabolism;John Wiley&Sons:New Jersey,1983;p 401.
(13) Hassan,J.;Sevignon,M.;Gozzi,C.;Schulz,E.;Lemaire,M.Chem.Rev.2002,102,1359.doi:10.1021/cr000664r
(14) Miyaura,N.;Suzuki,A.Chem.Rev.1995,95,2457.doi:10.1021/cr00039a007
(15) Bringmann,G.;Mortimer,A.J.P.;Keller,P.A.;Gresser,M.J.;Garner,J.;Breuning,M.Angew.Chem.Int.Edit.2005,44,5384.
(16) Kirsch,P.;Bremer,M.Angew.Chem.Int.Edit.2000,39,4216.
(17) Bauer,E.B.Curr.Org.Chem.2008,12,1341.doi:10.2174/138527208786241556
(18) Bolm,C.;Legros,J.;Paih,J.L.;Zani,L.Chem.Rev.2004,104,6217.doi:10.1021/cr040664h
(19)Czaplik,W.M.;Mayer,M.;Cvengros,J.;von Wangelin,A.J.ChemSusChem2009,2,396.doi:10.1002/cssc.v2:5
(20) Sherry,B.D.;Furstner,A.Accounts Chem.Res.2008,41,1500.doi:10.1021/ar800039x
(21) Furstner,A.;Leitner,A.Angew.Chem.Int.Edit.2002,41,609.
(22) Furstner,A.;Leitner,A.;Mendez,M.;Krause,H.J.Am.Chem.Soc.2002,124,13856.doi:10.1021/ja027190t
(23)Correa,A.;Mancheno,O.G.;Bolm,C.Chem.Soc.Rev.2008,37,1108.doi:10.1039/b801794h
(24) Furstner,A.;Martin,R.Chem.Lett.2005,34,624.doi:10.1246/cl.2005.624
(25) Furstner,A.;Martin,R.;Krause,H.;Seidel,G.;Goddard,R.;Lehmann,C.W.J.Am.Chem.Soc.2008,130,8773.doi:10.1021/ja801466t
(26)Noda,D.;Sunada,Y.;Hatakeyama,T.;Nakamura,M.;Nagashima,H.J.Am.Chem.Soc.2009,131,6078.doi:10.1021/ja901262g
(27) Scheiper,B.;Bonnekessel,M.;Krause,H.;Furstner,A.J.Org.Chem.2004,69,3943.doi:10.1021/jo0498866
(28) Molander,G.A.;Rahn,B.J.;Shubert,D.C.Tetrahedron Lett.1983,24,5449.doi:10.1016/S0040-4039(00)94109-1
(29) Quintin,J.;Franck,X.;Hocquemiller,R.;Figadere,B.Tetrahedron Lett.2002,43,3547.doi:10.1016/S0040-4039(02)00568-3
(30) Hocek,M.;Dvorakova,H.J.Org.Chem.2003,68,5773.doi:10.1021/jo034351i
(31)Hatakeyama,T.;Nakamura,M.J.Am.Chem.Soc.2007,129,9844.doi:10.1021/ja073084l
(32) Hatakeyama,T.;Hashimoto,S.;Ishizuka,K.;Nakamura,M.J.Am.Chem.Soc.2009,131,11949.doi:10.1021/ja9039289
(33)Mayer,M.;Welther,A.;von Wangelin,A.J.ChemCatChem2011,3,1567.doi:10.1002/cctc.v3.10
(34)Clayton,H.S.;Moss,J.R.;Dry,M.E.J.Organomet.Chem.2003,688,181.doi:10.1016/j.jorganchem.2003.08.044
(35) Knolker,H.J.Chem.Soc.Rev.1999,28,151.doi:10.1039/a705401g
(36) Gulak,S.;von Wangelin,A.J.Angew.Chem.Int.Edit.2012,51,1357.doi:10.1002/anie.201106110
(37) Smith,R.S.;Kochi,J.K.J.Org.Chem.1976,41,502.doi:10.1021/jo00865a019
(38)Bogdanovic,B.;Schwickardi,M.Angew.Chem.Int.Edit.2000,39,4610.
(39) Kleimark,J.;HedstromA.;Larsson P.F.;Johansson,C.;Norrby,P.ChemCatChem2009,1,152.doi:10.1002/cctc.v1:1
(40) Ren,Q.;Guan,S.;Jiang,F.;Fang,J.J.Phys.Chem.A2013,117,756.doi:10.1021/jp3045498
(41)Czaplik,W.M.;Mayer,M.;von Wangelin,A.J.ChemCatChem2011,3,135.doi:10.1002/cctc.201000276
(42) Mo,Z.;Zhang,Q.;Deng,L.Organometallics2012,31,6518.doi:10.1021/om300722g
(43) Becke,A.D.Phys.Rev.A1988,38,3098.doi:10.1103/PhysRevA.38.3098
(44) Becke,A.D.J.Chem.Phys.1993,98,5648.doi:10.1063/1.464913
(45) Hertwig,R.H.;Koch,W.Chem.Phys.Lett.1997,268,345.doi:10.1016/S0009-2614(97)00207-8
(46)Lee,C.T.;Yang,W.T.;Parr,R.G.Phys.Rev.B1988,37,785.doi:10.1103/PhysRevB.37.785
(47) Stephens,P.J.;Devlin,F.J.;Chabalowski,C.F.;Frisch,M.J.J.Phys.Chem.1994,98,11623.doi:10.1021/j100096a001
(48) Vosko,S.H.;Wilk,L.;Nusair,M.Can.J.Phys.1980,58,1200.doi:10.1139/p80-159
(49) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03,Revision C.02;Gaussian Inc.;Wallingford,CT,2004.
(50) Krishnan,R.;Binkley,J.S.;Seeger,R.;Pople,J.A.J.Chem.Phys.1980,72,650.doi:10.1063/1.438955
(51) McLean,A.D.;Chandler,G.S.J.Chem.Phys.1980,72,5639.doi:10.1063/1.438980
(52)Andrae,D.;Haussermann,U.;Dolg,M.;Stoll,H.;Preuss,H.Theor.Chim.Acta.1990,77,123.doi:10.1007/BF01114537
(53)Cossi,M.;Rega,N.;Scalmani,G.;Barone,V.J.Comput.Chem.2003,24,669.doi:10.1002/jcc.10189
(54) Castejon,H.;Wiberg,K.B.J.Am.Chem.Soc.1999,121,2139.doi:10.1021/ja983736t
(55)Perng,B.C.;Newton,M.D.;Raineri,F.O.;Friedman,H.L.J.Chem.Phys.1996,104,7153 and 7177.
(56) Najafi,M.;Zahedi,M.;Klein,E.Comput.Theor.Chem.2011,978,16.doi:10.1016/j.comptc.2011.09.014
(57) Schubert,G.;Papai,I.J.Am.Chem.Soc.2003,125,14847.doi:10.1021/ja035791u
(58) Belelli,P.G.;Damiani,D.E.;Castellani,N.J.Chem.Phys.Lett.2005,401,515.doi:10.1016/j.cplett.2004.11.089
(59) Feng,J.;Ren,Q.H.Acta.Phys.-Chim.Sin.2014,30,821.[蒋 峰,任清华.物理化学学报,2014,30,821.]
(60) Nova,A.;Ujaque,G.;Maseras,F.;Liedos,A.;Espinet,P.J.Am.Chem.Soc.2006,128,14571.doi:10.1021/ja0635736
猜你喜欢
中南民族大学学报(自然科学版)(2022年6期)2022-11-02
分子催化(2022年1期)2022-11-02
上海大学学报(自然科学版)(2021年2期)2021-02-26
上海大学学报(自然科学版)(2020年6期)2021-01-11
中南民族大学学报(自然科学版)(2020年6期)2020-12-22
上海大学学报(自然科学版)(2020年4期)2020-05-24
———理学院
西安航空学院学报(2018年1期)2018-02-05
杭州师范大学学报(自然科学版)(2017年3期)2017-06-15
中国塑料(2015年10期)2015-10-14
浙江科技学院学报(2012年4期)2012-11-05
