
脊髓小脑共济失调基因分型、临床及影像学特征分析*
2015-02-23 10:56夏明荣,黄月,张杰文等
郑州大学学报(医学版) 2015年6期
关键词:临床特征
脊髓小脑共济失调基因分型、临床及影像学特征分析*
夏明荣,黄月#,张杰文,李书剑
郑州大学人民医院(河南省人民医院)神经内科 郑州 450003
关键词脊髓小脑共济失调;三核苷酸重复;基因分型;临床特征
脊髓小脑共济失调(spinocerebellar ataxia,SCA)是一组具有临床和遗传异质性的神经系统遗传疾病[1-2],多呈常染色体显性遗传,主要表现为慢性进行性加重的肢体共济失调、构音障碍及眼球运动障碍,影像学研究[1-4]表明病变主要累及大脑、小脑、脑干及脊髓。目前报道[1,5-6]的SCA亚型多达40种,其发病年龄及临床症状多数相互重叠,因此仅仅依据临床表型特征及影像学评估对患者诊断分型较为困难,而基因检测则有助于明确诊断。该研究主要对临床诊断为SCA的一家系9例患者及有血缘关系、临床表现正常的27例家系成员进行基因检测,以确定基因型,同时对患者的临床特点进行总结分析,为临床诊断提供相关资料。
1对象与方法
1.1研究对象该家系先证者在河南省人民医院神经内科临床诊断为SCA,诊断参考Harding[7]标准。通过对该家系进行详细调查,6代成员中共27名发病者,其中男12例,女15例;符合常染色体显性遗传,绘制家系图(图1)。现存9例患者中男6例,女3例,年龄34~59(46.3±8.8)岁,发病年龄30~49(38.8±8.3)岁;27例临床表现正常的家系成员中男14例,女13例,年龄4~40(20.6±11.0)岁。在采血前均告知该研究目的,签署知情同意书,对患者及部分成员进行详细的体格检查。
1.2目的基因的提取、扩增及检测常规抽取该家系患者及其健康成员外周静脉血5 mL,38 g/L枸橼酸钠抗凝, 采用血液DNA提取试剂盒(北京天根生物工程公司)提取DNA作为PCR模板。根据文献[8-10]设计合成引物(上海生工生物工程股份有限公司)。PCR反应体系(50 μL):Premix Tag 25 μL,上、下游引物各2 μL(10 μmol/L),模板DNA 5 μL,然后加入ddH2O至总体积50 μL。PCR反应条件:94 ℃预变性5 min;94 ℃变性30 s,退火温度根据引物不同有所不同,72 ℃延伸30 s;72 ℃再延伸7 min(表1)。扩增反应在ABI9700扩增仪(美国ABI公司)中进行。PCR产物以80 g/L变性聚丙烯酰胺凝胶进行电泳检测鉴定。
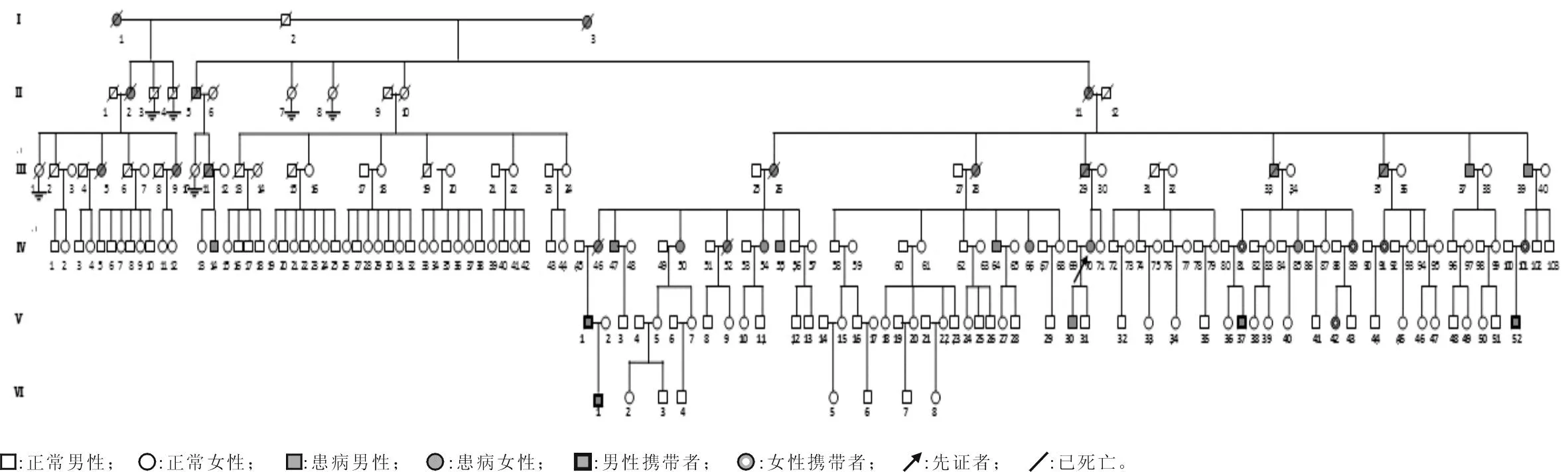
图1 SCA3型家系图(符合常染色体显性遗传模式)
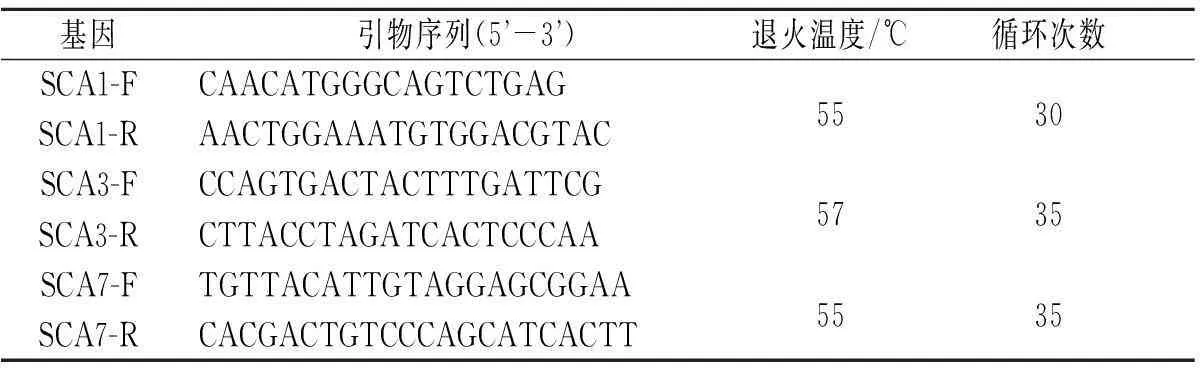
基因引物序列(5 -3 )退火温度/℃循环次数SCA1-FCAACATGGGCAGTCTGAG5530SCA1-RAACTGGAAATGTGGACGTACSCA3-FCCAGTGACTACTTTGATTCG5735SCA3-RCTTACCTAGATCACTCCCAASCA7-FTGTTACATTGTAGGAGCGGAA5535SCA7-RCACGACTGTCCCAGCATCACTT
F:上游引物;R:下游引物。
2结果
2.1临床特征9例患者虽来自同一家系,但临床表现有所不同。其中行走不稳、言语含糊9例(且为主要临床表现),饮水呛咳8例,悬雍垂右偏1例,悬雍垂左偏1例,复视6例,水平眼震7例,慢眼扫视3例,肌张力增高2例,腱反射亢进4例,不自主震颤5例,踝阵挛阳性6例,下肢肌肉萎缩4例,病理征7例,认知功能障碍3例,自主神经功能障碍3例。先证者(Ⅳ-70)及其叔叔(Ⅲ-37)头颅MRI检查示小脑及其蚓部萎缩,且Ⅲ-37双侧颞叶不对称性萎缩。先证者儿子(Ⅴ-30)头颅MRI检查未见明显异常。
2.2PCR结果见图2。所有受检者SCA1、SCA7目的基因PCR产物电泳结果显示一条带,片段大小分别为210 bp、270 bp,在正常范围,说明该家系患者及健康成员2条等位基因均无SCA1、SCA7基因CAG三核苷酸重复扩增。9例患者及9例家系“健康”成员SCA3基因扩增电泳均显示两条带,其中一条大小在430 bp左右(正常条带大小在250 bp左右),明显高于正常条带,提示9例患者及9例健康成员SCA3基因内其中一条染色体存在CAG三核苷酸重复扩增。9例患者进行基因检测前已发病,临床可确诊为SCA3型;9例“健康”家系成员无明显临床表现,根据PCR产物电泳结果,确诊为症状前患者。
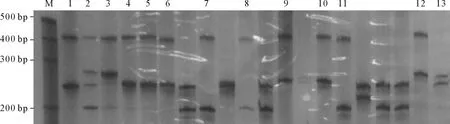
图2 该家系SCA3基因PCR扩增产物电泳图(部分)
M:Marker;1~13:分别为Ⅲ-37、Ⅲ-39、Ⅳ-47、Ⅳ-50、Ⅳ-55、Ⅳ-64、Ⅳ-70(先证者)、Ⅳ-81、Ⅳ-85、Ⅳ-89、Ⅳ-91、阳性对照及阴性对照。其中Ⅲ-37、Ⅲ-39、Ⅳ-47、Ⅳ-50、Ⅳ-55、Ⅳ-64、Ⅳ-70和Ⅳ-85为该家系患者,Ⅳ-81、Ⅳ-89和Ⅳ-91为症状前患者,其余为家系健康个体。
3讨论
SCA是一组遗传性神经退行性疾病,各亚型临床表现虽有重叠,但其表现仍具有高度的异质性,根据致病基因或致病位点的不同可将其分为40余种亚型,其中SCA1、SCA2、SCA3、SCA6、SCA7、SCA17及DRPLA发病原因均是致病基因编码区特定片段CAG三核苷酸重复扩增,编码产生异常的多聚谷氨酰胺链所致,因此该类疾病又可称为多聚谷氨酰胺疾病[11]。流行病学研究发现,SCA3型在世界范围内是最常见的亚型[12],占SCA的20%~50%[13],但其发病率在不同国家、地区及种族之间具有一定的差异。SCA3型又称马查多-约瑟夫病(Machado-Joseph diease,MJD),其致病基因定位于14q32.12, 含有11个外显子,编码致病蛋白ataxin-3/MJDp[14],基因突变是第10号外显子不稳定的CAG三核苷酸重复扩增。为了节省实验时间及提高效率,作者首先对该家系进行了SCA3、SCA1及SCA7的基因检测。
SCA1发病多在30岁以后,中青年发病,且病程进展较其他亚型快[15]。临床表现主要为进行性的平衡及协作功能的丧失,认知功能障碍,凝视麻痹,慢眼扫视,构音障碍,吞咽困难,周围神经病,锥体系及锥体外系症状;运动和感觉神经传导速度中度减慢,视觉诱发电位延迟,躯体感觉诱发电位丧失,运动诱发电位丧失[16];头颅MRI可见尾状核、豆状核、小脑及脑干的萎缩。SCA7发病年龄变化较大,儿童时期至76岁不等,平均发病年龄20岁,病程进展缓慢[17]。临床表现为小脑性共济失调,黄斑及视网膜变性,慢眼扫视,眼外肌麻痹,吞咽困难,躯体感觉及神经精神症状,其中黄斑及视网膜变性为主要特征[17];神经传导速度未见明显异常,眼电图示视觉功能障碍[16];MRI示小脑和脑干萎缩。SCA3的发病年龄从5岁至75岁不等,大多数常见病例为中青年发病,临床主要以进行性加重的小脑共济失调,腱反射亢进或消失,周围神经、肌肉萎缩,帕金森样症候群,肌张力障碍、肌肉痉挛为特征,其他症状如眼外肌麻痹、面肌及舌肌的肌纤维震颤,眼球突出也可以作为临床诊断SCA3的依据[18];头颅MRI表现为第四脑室扩大,大脑半球皮层、脑桥、基底核、小脑蚓部的中度萎缩[19]。
该研究中9例患者经基因检测确诊为SCA3型,其发病年龄和临床表现符合文献[12]报道的SCA3型患者的临床特征。9例患者随着病程的进展均出现不自主的肢体震颤及阵挛,且发病年龄越早,疾病进展越快,阵挛出现越早,且可能先于其他症状出现;先证者儿子(V-30),15岁发病,病程3 a,发病症状同其他患者,但出现明显的不自主震颤及阵挛,且伴有左下肢肌肉轻度萎缩,头颅MRI平扫未见明显异常。先证者叔叔(Ⅲ-37)病程10 a,基本丧失生活能力,高级皮层功能减退,头颅MRI示小脑轻度萎缩,大脑皮层及颞叶不对称萎缩,结合患者临床表现,不排除患者认知障碍可能。据报道[20]大多数SCA3型患者不出现认知功能障碍,但是也有研究[21]发现SCA3型患者晚期可以出现痴呆。先证者(Ⅳ-70)除上述症状外,伴有月经不规律,其儿子(V-30)易出汗,考虑有自主神经功能受损。
通过对该家系患者的临床及影像特征总结分析,结合基因分型,在一定程度上补充了SCA3疾病谱的临床特征,为该病早期诊断提供相应依据。由于该家系人员较多,尚未对参与基因检测者的CAG三核苷酸重复次数进行检测,后续工作将进一步对该家系成员基因内CAG重复在该家系各代成员之间的变化以及与发病年龄、病程、疾病严重程度的关系进行研究;同时对该家系症状前患者进行长期随访追踪。目前针对该病尚无有效的治疗和预防方法,对于已发病者主要进行对症治疗,而对症状前患者,需及早进行干预以减轻症状,延长存活时间。
参考文献
[1]Chen JW,Zhao L,Zhang F,et al.Clinical characteristics, radiological features and gene mutation in 10 Chinese families with spinocerebellar ataxias[J].Chin Med J(Engl),2015,128(13):1714
[2]吴英,魏倩倩,商慧芳.脊髓小脑共济失调基因型分布及临床特点分析[J].中国实用内科杂志,2014,34(5):512
[3]Ramachandra NB,Kusuma L.An understanding of spinocerebellar ataxia[J].Indian J Med Res,2015,141(2):148
[4]吴丹丹,刘小璇,刘建元,等.一个脊髓小脑共济失调家系的分子遗传学研究[J].解放军医学杂志,2015,40(8):638
[5]Serrano-Munuera C,Corral-Juan M,Stevanin G,et al.New subtype of spinocerebellar ataxia with altered vertical eye movements mapping to chromosome 1p32[J].JAMA Neurol,2013,70(6):764
[6]Tsoi H,Yu AC,Chen ZS,et al.A novel missense mutation in CCDC88C activates the JNK pathway and causes a dominant form of spinocerebellar ataxia[J].J Med Genet,2014,51(9):590
[7]Harding AE.Clinical features and classification of inherited ataxias[J].Adv Neurol,1993,61(1):1
[8]Lin JX,Ishikawa K,Sakamoto M,et al.Direct and accurate measurement of CAG repeat configuration in the ataxin-1 (ATXN-1) gene by "dual-fluorescence labeled PCR-restriction fragment length analysis"[J].J Hum Genet,2008,53(4):287
[9]Bettencourt C,Santos C,Kay T,et al.Analysis of segregation patterns in Machado-Joseph disease pedigrees[J].J Hum Genet,2008,53(10):920
[10]Lin Y,Zheng JY,Jin YH,et al.Trinucleotide expansions in the SCA7 gene in a large family with spinocerebellar ataxia and craniocervical dystonia[J].Neurosci Lett,2008,434(2):230
[11]Fan HC,Ho LI,Chi CS,et al.Polyglutamine(PolyQ) diseases: genetics to treatments[J].Cell Transplant,2014,23(4/5):441
[12]Jacobi H,Minnerop M,Klockgether T.The genetics of spinocerebellar ataxias[J].Nervenarzt,2013,84(2):137
[13]Wang J,Shen L,Lei L,et al.Spinocerebellar ataxias in mainland China: an updated genetic analysis among a large cohort of familial and sporadic cases[J].Zhong Nan Da Xue Xue Bao Yi Xue Ban,2011,36(6):482
[14]Evers MM,Tran HD,Zalachoras I,et al.Ataxin-3 protein modification as a treatment strategy for spinocerebellar ataxia type 3: removal of the CAG containing exon[J].Neurobiol Dis,2013,58:49
[15]Donato SD,Mariotti C,Taroni F.Spinocerebellar ataxia type 1[J].Handb Clin Neurol,2012,103:399
[16]Rüb U,Schöls L,Paulson H,et al.Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxias type 1,2,3,6 and 7[J].Prog Neurobiol,2013,104:38
[17]Donis KC,Mattos EP,Silva AA,et al.Infantile spinocerebellar ataxia type 7:case report and a review of the literature[J].J Neurol Sci,2015,354(1/2):118
[18]]Jung BC,Choi SI,Du AX,et al.MRI shows a region-specific pattern of atrophy in spinocerebellar ataxia type 2[J].Cerebellum,2012,11(1):272
[19]Ma J,Wu C,Lei J,et al.Cognitive impairments in patients with spinocerebellar ataxia types 1, 2 and 3 are positively correlated to the clinical severity of ataxia symptoms[J].Int J Clin Exp Med,2014,7(12):5765
[20]Fancellu R,Paridi D,Tomasello C,et al.Longitudinal study of cognitive and psychiatric functions in spinocerebellar ataxia types 1 and 2[J].J Neurol,2013,260(12):3134
[21]Roeske S,Filla I,Heim S,et al.Progressive cognitive dysfunction in spinocerebellar ataxia type 3[J].Mov Disord,2013,28(10):1435
doi:10.13705/j.issn.1671-6825.2015.06.034
通信作者#,女,1965年10月生,博士,主任医师,研究方向:神经系统变性病,E-mail:hy139378@163.com
猜你喜欢
上海医药(2016年23期)2016-12-22
中国现代医生(2016年27期)2016-12-21
中国实用医药(2016年28期)2016-12-07
科教导刊·电子版(2016年26期)2016-11-21
中国实用医药(2016年26期)2016-11-07
中国实用医药(2016年22期)2016-08-19
中国实用医药(2016年19期)2016-08-05
中国实用医药(2016年17期)2016-07-26
中国实用医药(2016年15期)2016-05-24
中国实用医药(2016年10期)2016-05-04
