
一例罕见的新的TCIRG1基因杂合性突变引起的婴儿恶性石骨症*
2015-05-16 00:50曾秉辉胡悦林景象一张永玲王一鸣中山大学中山医学院医学遗传学教研室疾病基因组研究所广东广州50080广州市妇女儿童医疗中心影像科优生围产研究所广东广州506
中国病理生理杂志 2015年7期
胡 彬,曾秉辉,胡悦林,赵 强,景象一,张永玲,王一鸣△(中山大学中山医学院医学遗传学教研室,疾病基因组研究所,广东广州50080;广州市妇女儿童医疗中心影像科,优生围产研究所,广东广州506)
一例罕见的新的TCIRG1基因杂合性突变引起的婴儿恶性石骨症*
胡彬1,2,曾秉辉1,2,胡悦林3,赵强1,2,景象一4,张永玲4,王一鸣1,2△
(中山大学1中山医学院医学遗传学教研室,2疾病基因组研究所,广东广州510080;广州市妇女儿童医疗中心3影像科,4优生围产研究所,广东广州510623)
[摘要]目的:研究1例婴儿恶性石骨症患者的致病基因及其突变。TCIRG1和CLCN7是婴儿恶性石骨症最常见的致病基因。前者被认为是纯合性致病基因,国外只有6例其杂合性改变也致本病的报导,而我国无杂合性突变导致本病的报道。方法:通过骨组织X线检查结合临床症状及体征确诊1例散发性婴儿恶性石骨症患者。提取患者及其父母的外周血基因组DNA,PCR扩增TCIRG1和CLCN7基因外显子及其剪切位点序列,对PCR产物直接测序。用TCRIG1基因附近的微卫星标记和SNP构建患者及其父母的单倍型。用染色体微阵列分析技术对患者及其母亲进行TCIRG1基因拷贝数目变异的检测。结果:患者TCIRG1基因第5号外显子内发现一个4个碱基的缺失突变c.449_452delAGAG( p.Gln149Glnfs16),该突变使得基因3’端编码的666个氨基酸被截断,失去了整个ATP酶V0复合结构域。患儿双亲TCIRG1和CLCN7基因的突变检测及单倍体构建证实该突变来源于患者父亲。染色体微阵列分析未发现患儿及其母亲携带有任何累及TCIRG1及CLCN7基因的拷贝数目变异。结论:本研究发现了1例TCIRG1基因新的杂合性突变所致的婴儿恶性石骨症。这是我国TCIRG1基因杂合性突变引起婴儿恶性石骨症的首例报道。这个发现可用于婴儿恶性石骨症的分子诊断。
[关键词]婴儿恶性石骨症; TCIRG1基因;缺失突变
婴儿恶性石骨症( infantile malignant osteopetrosis,IMO)是一种罕见的单基因遗传性疾病,是石骨症最严重的一种类型[1]。该病骨骼的异常表现为骨密度增高、病理性骨折、骨髓炎,其它临床表现还有全血细胞减少、髓外造血、肝脾肿大、第II、VII、VIII对颅神经受压、脑积水、低钙血症等[2]。T细胞免疫调节子1( T-cell immune regulator 1,TCIRG1)和氯离子通道7( chloride channel 7,CLCN7)是最常见的致病基因[3-4]。TCIRG1基因编码V型ATP酶116 kD同工型a3( V-ATPase 116 kD isoform a3)[5],CLCN7基因编码氯离子通道蛋白7( chloride channel protein 7,ClC-7)[6]。TCIRG1一般被认为只在纯合性突变或复合杂合性突变时致病,但国外有3个学者报道了6例其杂合性突变所致石骨症[7-9]。而在我国还未存在类似文献。本研究对1例石骨症患者进行TCIRG1基因突变筛查,现报道如下。
材料和方法
1研究对象
本研究中的石骨症病人来自于广州市妇女儿童医疗中心。本研究得到了患者及其家属的知情同意和中山大学中山医学院医学伦理委员会的批准。
患者2岁时因摔伤到广州市妇女儿童医疗中心检查,确诊为石骨症。牙齿萌出障碍,至7岁时仍只萌出4颗牙齿。视力受损,眼底镜检查显示视神经萎缩。血常规显示红细胞计数为3. 29×1012/L(正常范围3. 79×1012~5. 88×1012/L),血红蛋白浓度为77 g/L(正常范围110~147 g/L)。腹部B超显示肝脾肿大。脑电图无明显异常。
头部CT及X线片(图1)显示颅骨广泛硬化、增厚,板障间隙消失。全身X线片显示脊柱椎体上下缘致密增厚,中间较稀疏,呈“夹心饼”样改变。盆骨骨髓腔消失,呈现“骨中骨”的特殊表现。所摄诸长骨骨密度增高,骨髓腔消失,干骺端呈“烧瓶样”改变。
2方法
2.1抽提基因组DNA取患者及其父母外周血3 mL,乙二胺四乙酸( ethylene diamine tetraacetic acid,EDTA)抗凝,按天根血液基因组提取试剂盒操作指南提取DNA。
2.2引物设计和PCR从UCSC数据库( http: / / genome.ucsc.edu)获取TCIRG1和CLCN7基因序列,用Oligo 6. 0软件分别设计10和16对引物,覆盖所有编码区域和剪切位点。这些引物由深圳华大基因研究院合成。PCR扩增反应体系为30 μL体系: 2× GCI缓冲液15 μL,2. 5 mmol/L dNTP混合物3 μL,LA Taq DNA聚合酶0. 3 μL,3. 2 μmol/L上、下游引物各1 μL,DNA模板1 μL,灭菌水补足体积至30 μL。反应条件: 95℃3 min; 95℃30 s,58℃45 s,72 ℃1 min,40个循环; 72℃10 min。
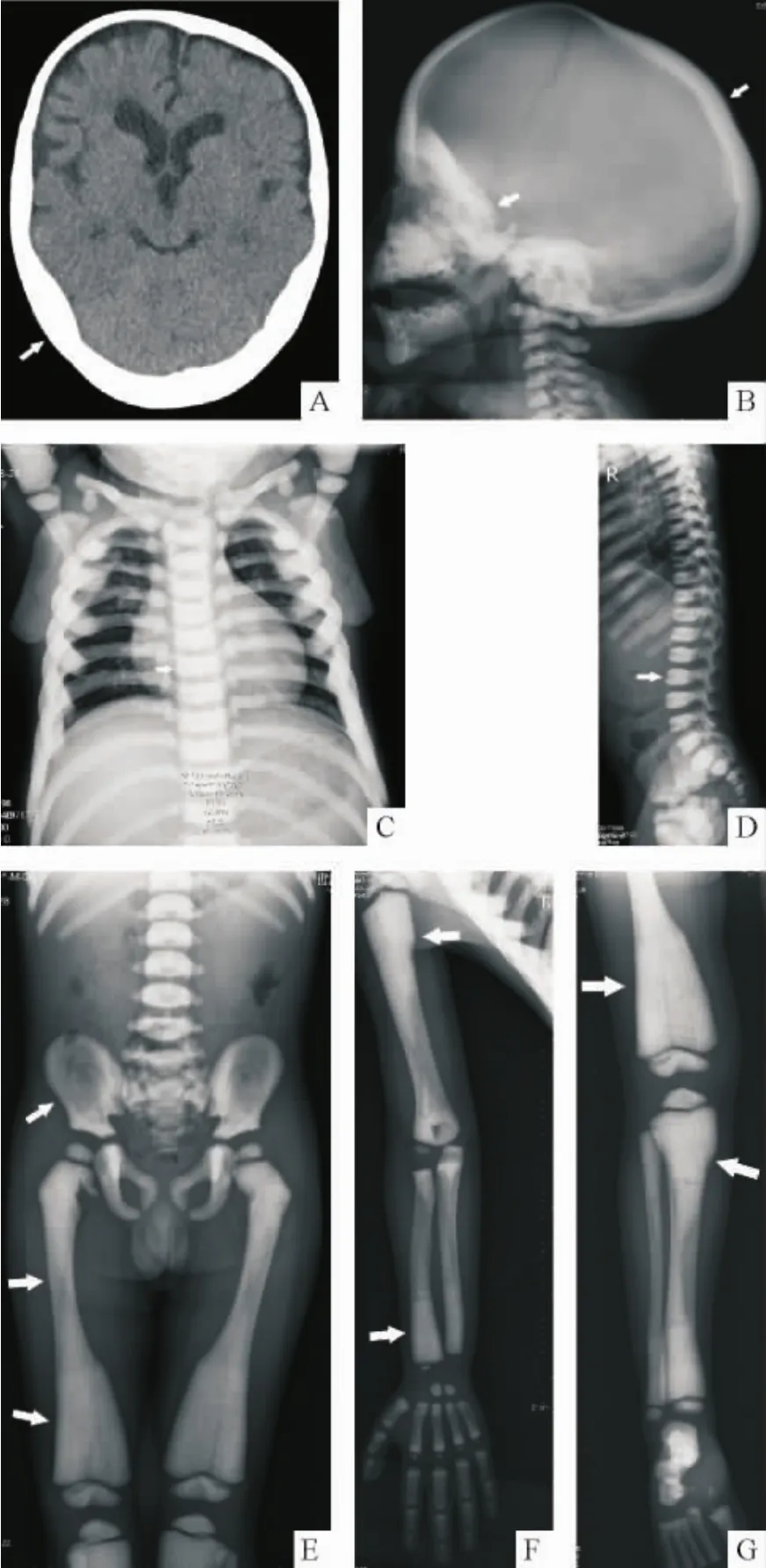
Figure 1.Radiological images of the patient.A,B: skull was generally sclerotic and the thickness increased; C,D: vertebral endplates was sclerotic and resulted in “sandwich vertebrae”appearance; E~G: the long bones showed the flask deformity and absence of bone marrow cavity; the bone marrow cavity of pelvis disappeared and showed“bone in bone”appearance.图1 患者的影像学资料
2.3Sanger测序及突变鉴定在ABI 3730XL DNA测序仪上对PCR产物进行直接测序。测序结果用Sequence Scanner分析,并与UCSC数据库基因参考序列比对,突变命名采用标准命名法( http: / /www.hgvs.org/mutnomen/),+ 1代表翻译起始密码子ATG中的A。对检测到的突变,用PCR法扩增父母相应的外显子,并用直接测序法测序分析。
2.4基因分型及单倍体构建我们对患者及其父母TCIRG1基因附近的微卫星标记D11S987、D11S4162和D11S1314进行PCR扩增,并在ABI 3730XL DNA测序仪上进行基因型鉴定。在TCIRG1基因内及其附近选取杂合度较高的SNP rs4147780、rs12273861、rs2075609和rs11481设计4对引物行PCR扩增,并用直接测序法鉴定其基因型。对rs7116924、rs3808974、rs10896289和rs11228127,则分别在第1、3、5和10对引物的测序结果中分析其基因型。根据所有基因型信息构建单倍体。
2.5染色体微阵列分析我们用CytoScan 750K Array( Affymetrix)对患者及其母亲进行染色体微阵列分析。我们根据Affymetrix的标准操作流程进行了DNA提取、扩增、片段化、标记和杂交。实验结果用Chromosome Analysis Suite软件进行分析。
结果
1 TCIRG1基因
患者TCIRG1基因直接测序结果显示,第5个外显子内检测到1个小的杂合缺失突变c.449 _ 452delAGAG( p.Gln149Glnfs16),见图2。该突变使得开放阅读框改变,在肽链第164位引入终止密码子,使得3’端666个氨基酸被截断,失去了整个ATP 酶V0复合结构域,见图3。由于该结构域是V型ATP酶116 kD同工型a3蛋白实现氢离子跨膜转运的结构基础,因此可以预测患者的突变使该蛋白完全失去功能。对患儿双亲的突变检测及单倍体构建结果表明该突变遗传自患者父亲,见图4。
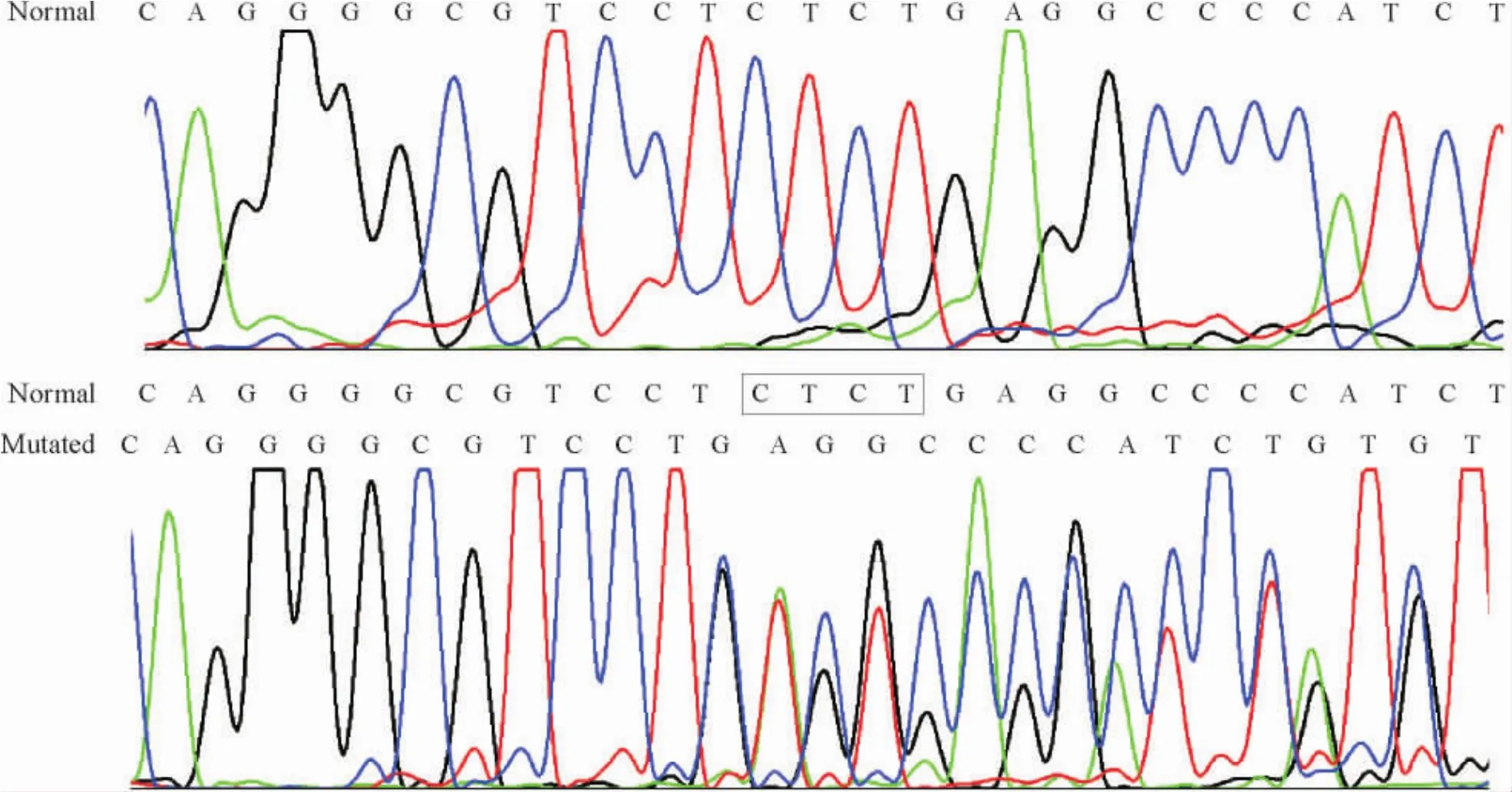
Figure 2.Partial reverse sequence of the patient ( lower) and his mother ( upper).The c.449_452delAGAG ( p.Gln149Glnfs16) mutation of the patient was showed in the box.图2 患者和其母亲的部分反向测序图
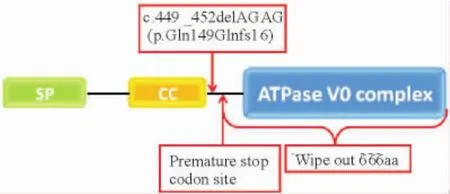
Figure 3.Prediction of the effect of c.449_452delAGAG ( p.Gln149Glnfs16) mutation.The mutation was predicted to wipe out 666 amino acids from the V-ATPase 116 kD isoform a3 protein.SP: signal peptide motif; CC: coiled-coil motif.图3 c.449_452delAGAG( p.Gln149Glnfs16)突变效果预测
2 CLCN7基因
对CLCN7基因的筛查未发现致病性突变。
3染色体微阵列分析
染色体微阵列分析结果未发现患者及其母亲累及TCIRG1及CLCN7基因的拷贝数目变异( CNV,copy number variation)。
讨论
TCIRG1基因及CLCN7基因是最常见的婴儿恶性型石骨症的致病基因,前者被认为是隐性基因。我们在患者中发现了TCIRG1基因的新的杂合性突变,而CLCN7基因未发现异常。这一新的TCIRG1基因突变c.449_452delAGAG( p.Gln149Glnfs16)造成了氨基酸的移码改变,并截断了V型ATP酶116 kD同工型a3蛋白,使其失去必不可少的ATP酶V0复合结构域,因此是一个致病性突变。由于Sanger测序不能检测拷贝数目变异[10-11],因此我们对患者及其母亲进行染色体微阵列分析。染色体微阵列研究结果排除了累及本基因的拷贝数目变异。这表明,本基因的杂合性突变也可能引起本病,这种杂合性突变致病的报道国外已有[7-9],但在国内为首例。
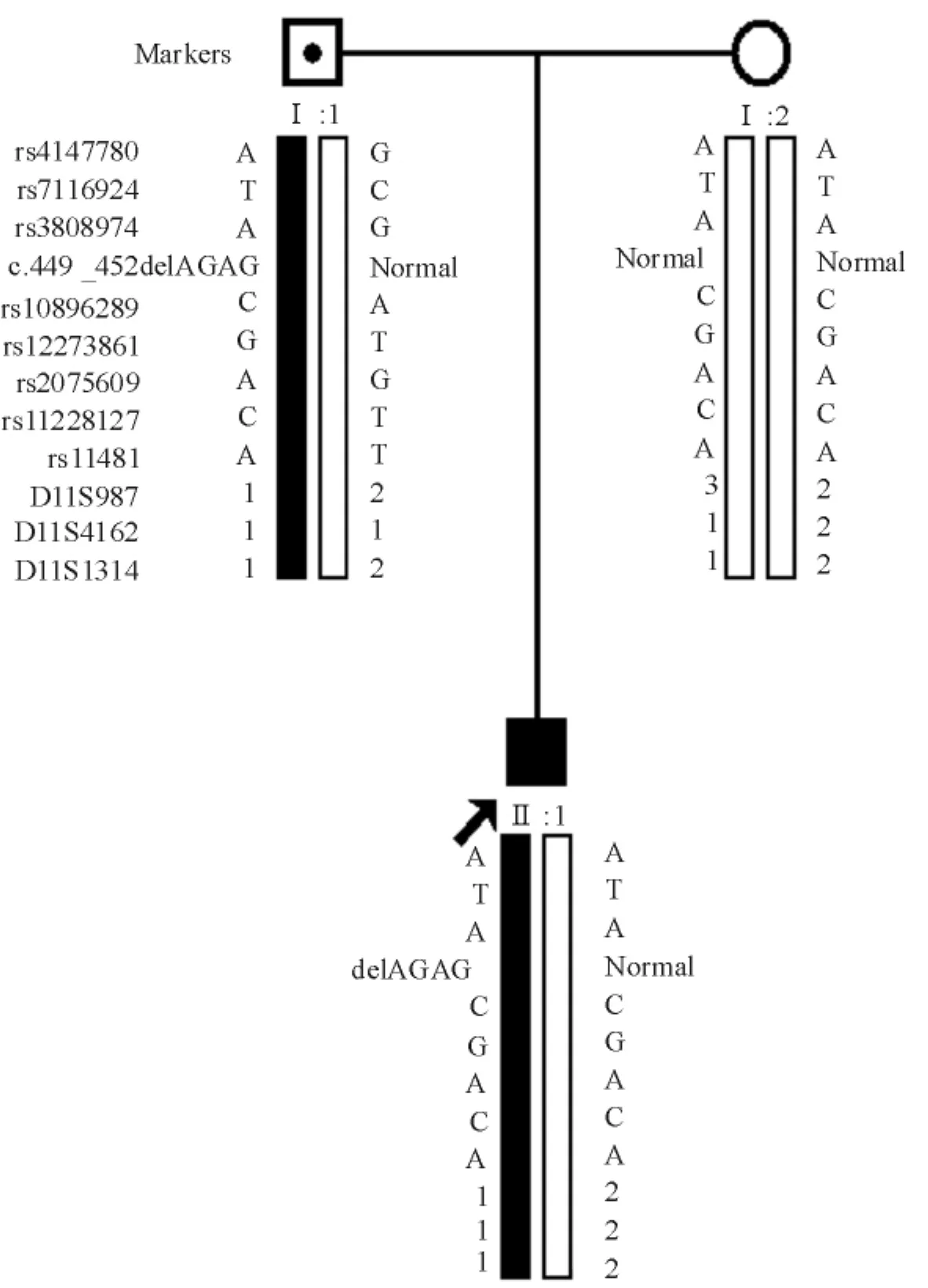
Figure 4.Pedigree of the family and haplotype analysis.Black bars depicted the disease-causing haplotypes.图4 家系图和单倍型分析
本文作者曾报道一例婴儿恶性型石骨症[12],该患者由TCIRG1基因c.242delC( p.Pro81ArgfsX85)突变和c.1114C>T( p.Gln372X)突变引起。其中c.242delC( p.Pro81ArgfsX85)突变与本文报道的c.449 _452delAGAG( p.Gln149Glnfs16)突变相似,也造成移码突变,使V型ATP酶116 kD同工型a3蛋白3’端666个氨基酸被截断,使整个ATP酶V0复合结构域丢失。其不同之处主要在于,本文报道的c.449_ 452delAGAG突变出现在卷曲螺旋( coiled coil)之后,没有破坏卷曲螺旋的结构,而c.242delC突变出现在卷曲螺旋之前,破坏了卷曲螺旋。虽然本文报道的突变c.449_452delAGAG未影响卷曲螺旋,但由于该突变已经使整个ATP酶V0复合结构域丢失,因此即使突变蛋白仍有表达,也无法行使正常功能。
TCIRG1基因编码的V型ATP酶116 kD同工型a3蛋白是破骨细胞吸收骨组织的结构基础。V型ATP酶可以在消耗ATP的同时向分泌性溶酶体泵入氢离子,之后,分泌性溶酶体被转运到破骨细胞的骨组织面,与细胞膜融合,在破骨细胞和骨组织之间形成皱褶缘,并使皱褶缘和骨组织之间的液体酸化,促使骨组织被吸收[1]。当TCIRG1基因的突变使a3蛋白失去功能时,V型ATP酶失去转运氢离子的功能,破骨细胞无法吸收骨组织。
破骨细胞吸收骨组织的功能丧失后,骨组织不能进行改建,骨髓腔消失,骨密度增加。患者髓腔内造血功能受到不同程度的破坏,可表现为全血细胞减少,也可仅红细胞减少。这促使髓外造血的发生,患者可表现为肝脾肿大。骨组织改建功能的丧失,也使得各出颅神经孔狭窄,脑神经受压,患者可表现为视神经损伤,听力损伤。另外,由于牙齿的萌出需要相应的牙槽骨及时被吸收,因此石骨症患者往往还有牙齿不能萌出的表现[2]。本研究中患者表现为骨密度增加,红细胞减少,肝脾肿大,视神经受压,牙齿萌出障碍,可见本研究中患者的临床表现是非常典型的。
我们对患者双亲TCIRG1基因的检测及单倍体分析提示患者父亲也携带这一致病性突变,但其表型完全正常,这一结果提示,本基因是否还有外显不全的可能?造成外显不全现象的原因非常复杂,修饰基因、等位基因的随机表达、表观遗传学的修饰以及环境因素、患者的年龄、性别等因素都可在一定程度上影响疾病的外显率[13]。这些影响基因突变效果的因素,在石骨症另外一个基因CLCN7中得到了充分体现。CLCN7基因的突变可引起婴儿恶性型、中间型、成人型石骨症[4]。其中,婴儿恶性型和中间型是由纯合或复合杂合性突变造成的,成人型是由杂合性突变造成的,且成人型石骨症存在外显不全的现象。这些现象使我们有理由推测,有某些修饰基因、表观遗传学修饰等因素,能使石骨症的表型加重或减轻,甚至引起外显不全的现象。因此,本研究中TCIRG1基因新突变c.449 _ 452delAGAG ( p.Gln149Glnfs16)在家系中的外显不全现象,也可能是通过这些机制产生的。
综上所述,本研究在1例婴儿恶性型石骨症患者TCIRG1基因中发现了一个新的杂合性突变,这是国内第1例,国际上第7例由TCIRG1基因杂合性突变引起的石骨症的报导。这为进一步揭示婴儿恶性型石骨症的发病机制提供了新的数据。
[参考文献]
[1]Sobacchi C,Schulz A,Coxon FP,et al.Osteopetrosis: genetics,treatment and new insights into osteoclast function[J].Nat Rev Endocrinol,2013,9( 9) : 522-536.
[2]Stark Z,Savarirayan R.Osteopetrosis[J].Orphanet J Rare Dis,2009,4: 5.
[3]Scimeca JC,Quincey D,Parrinello H,et al.Novel mutations in the TCIRG1 gene encoding the a3 subunit of the vacuolar proton pump in patients affected by infantile malignant osteopetrosis[J].Hum Mutat,2003,21( 2) : 151-157.
[4]Frattini A,Pangrazio A,Susani L,et al.Chloride channel ClCN7 mutations are responsible for severe recessive,dominant,and intermediate osteopetrosis[J].J Bone Miner Res,2003,18( 10) : 1740-1747.
[5]Nishi T,Forgac M.The vacuolar ( H+) -ATPases: nature’s most versatile proton pumps[J].Nat Rev Mol Cell Biol,2002,3( 2) : 94-103.
[6]Kornak U,Kasper D,Bosl MR,et al.Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man [J].Cell,2001,104( 2) : 205-215.
[7]Wada K,Harada D,Michigami T,et al.A case of autosomal dominant osteopetrosis type II with a novel TCIRG1 gene mutation[J].J Pediatr Endocrinol Metab,2013,26 ( 5-6) : 575-577.
[8]Kornak U,Schulz A,Friedrich W,et al.Mutations in the a3 subunit of the vacuolar H+-ATPase cause infantile malignant osteopetrosis[J].Hum Mol Genet,2000,9( 13) : 2059-2063.
[9]Sobacchi C,Frattini A,Orchard P,et al.The mutational spectrum of human malignant autosomal recessive osteopetrosis[J].Hum Mol Genet,2001,10( 17) : 1767-1773.
[10]邓佳,邓伟平,钟诚,等.NF1基因新突变及我国首例NF1基因拷贝数目变异报道[J].中国病理生理杂志,2013,29( 2) : 320-323.
[11]郑灵燕,裴元元,李淼新,等.CCL3L1基因拷贝数目变异与慢性乙肝的易感性相关[J].中国病理生理杂志,2011,27( 10) : 1951-1955.
[12]Yuan P,Yue Z,Sun L,et al.Novel mutation of TCIRG1 and clinical pictures of two infantile malignant osteopetrosis patients[J].J Bone Miner Metab,2011,29( 2) : 251-256.
[13]Cooper DN,Krawczak M,Polychronakos C,et al.Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease[J].Hum Genet,2013,132 ( 10) : 1077-1130.
An unusual and novel heterozygous TCIRG1 mutation causes infantile malignant osteopetrosis
HU Bin1,2,ZENG Bing-hui1,2,HU Yue-lin3,ZHAO Qiang1,2,JING Xiang-yi4,ZHANG Yong-ling4,WANG Yi-ming1,2
(1Department of Medical Genetics,Zhongshan School of Medicine,2Institute of Disease Genomics,Sun Yat-sen University,Guangzhou 510080,China;3Department of Radiology,4Department of Prenatal Diagnostic Center,Guangzhou Women and Children’s Medical Center,Guangzhou 510623,China.E-mail: ywzhong@ hotmail.com)
[ABSTRACT]AIM: To investigate the underlying genetic changes of a Chinese patient with infantile malignant osteopetrosis ( IMO).IMO is a monogenic disease,mostly caused by mutations of TCIRG1 and CLCN7 genes.The former is believed a homozygous gene and only cause the disease in homozygous or compound heterozygous status.However,it has been reported that heterozygous mutations also cause the disease in 6 non-Chinese cases.METHODS: Genomic DNA was extracted from peripheral blood of the patient and his parents.All exons and splice sites of TCIRG1 and CLCN7 genes were amplified by PCR followed by Sanger sequencing.Mutation detection in the 2 genes was also investigated in the parents.Haplotypes were constructed by variations obtained in mutation detection and microsatillites flanking TCIRG1 gene in the family by Cyrillic.Chromosomal microarray analysis ( CMA) was performed to detect copy number variations ( CNV) of the patient and his mother.RESULTS: A novel mutation c.449_452delAGAG ( p.Gln149Glnfs16) was detected in the patient.This mutation truncated 666 amino acids at the C terminal of the V-ATPase 116 kD isoform a3 protein.It wiped out the entire ATPase V0 complex and was predicted to result in total loss of protein function.This mutation was also detected in the patient’s father.No pathogenic mutation was detected in CLCN7 gene.CMA did not reveal any CNV involving TCIRG1 or CLCN7 gene.CONCLUSION: We reported a novel heterozygous mutation of TCIRG1 gene causing IMO.This represents the first IMO case in China caused by heterozygous TCIRG1 gene mutation.
[KEY WORDS]Infantile malignant osteopetrosis; TCIRG1 gene; Deletion mutation
通讯作者△Tel: 020-87332055; E-mail: ywzhong@ hotmail.com
*[基金项目]国家自然科学基金资助项目( No.31471193)
[收稿日期]2015-01-12[修回日期]2015-02-27
[文章编号]1000-4718( 2015)07-1237-05
[中图分类号]R363
[文献标志码]A
doi:10.3969/j.issn.1000-4718.2015.07.015
