
十六烷基三甲基溴化铵插层氧化石墨结构的分子模拟
2015-06-01 10:45赵二正彭同江孙红娟姬广富
无机化学学报 2015年3期
赵二正 彭同江*,,2 孙红娟 刘 波 姬广富
(1西南科技大学固体废物处理与资源化教育部重点实验室,绵阳621010)
(2西南科技大学分析测试中心,绵阳621010)
(3中国工程物理研究院流体物理研究所冲击波物理与爆轰物理国防重点实验室,绵阳621900)
十六烷基三甲基溴化铵插层氧化石墨结构的分子模拟
赵二正1彭同江*,1,2孙红娟1刘 波1姬广富3
(1西南科技大学固体废物处理与资源化教育部重点实验室,绵阳621010)
(2西南科技大学分析测试中心,绵阳621010)
(3中国工程物理研究院流体物理研究所冲击波物理与爆轰物理国防重点实验室,绵阳621900)
利用分子模拟方法研究了十六烷基三甲基溴化铵(C16TAB)分子数对C16TAB/GO插层复合物的结构变化,探讨了C16TAB在GO层间的排列方式,并通过实验数据进行验证。模拟结果表明,优化后GO结构模型的层间距为0.849 nm;C16TAB/GO插层复合物的层间距随着C16TAB分子数的增加呈5个阶梯状逐渐增大,层间距分别为1.56、1.98、2.33、2.76和3.40 nm,插层饱和时C16TAB分子达到28个。实验结果显示,随着C16TAB分子数的增加,C16TAB/GO插层复合物的层间距逐渐增大,插层饱和时为3.40 nm,实验结果与模拟结果能够很好地吻合。C16TAB在GO层间可能的排列方式为1~5层平躺排列或单层平躺、单层倾斜和单层直立,从能量和结构的角度探明了C16TAB在GO层间的最优排列为1~5层平躺排列。
十六烷基三甲基溴化铵;氧化石墨;分子模拟;层间距;排列方式
氧化石墨(graphite oxide,GO)结构中含有大量的羟基(-OH)和环氧基(C-O-C)以及少量的羧基(-COOH)和羰基(-C=O)等含氧官能团[1-4],这些官能团在水溶液或碱水条件下容易发生电离,使GO的结构层表面带负电[5],可通过离子键作用力将阳离子型表面活性剂插入到GO层间,改善GO的亲油性,提高其在有机介质中的分散性及剥离效果。
季铵盐阳离子通过离子键作用优先与GO电离后的氧原子结合[6-7],之后随着季铵盐浓度的增大再通过疏水键作用[8-9]以分子吸附的方式插入GO层间,形成季铵盐/氧化石墨插层复合物。杨建国等[5]研究表明十六烷基三甲基溴化铵(C16TAB)的插入使GO表面的亲油性得到了改善。韩志东等[10]采用C16TAB有机改性GO的研究结果表明C16TAB有效地增大了GO的层间距离。Matsuo等[7]在研究季铵盐插层GO的有序化过程中提出,随着季铵盐浓度的增大,季铵盐在GO层间有单层平躺、双层平躺、单层倾斜、双层倾斜等不同的排列方式。Liu[11]、Matsuo[12]和林舜嘉等[13]的研究结果表明季铵盐/氧化石墨插层复合物的层间距随季铵盐链长的增长而增加,随季铵盐用量的增加而增大。目前,季铵盐在层状硅酸盐矿物插层方面的模拟计算研究已有诸多报道[14-18]。然而,关于季铵盐插层GO的研究集中于实验方面[5,7,10-13],且大部分的结构模型都是基于实验数据的一种半经验式推导所得,缺少理论的验证,对于季铵盐在GO层间排列方式及作用机理的分子模拟研究尚未见报道。
本文从分子模拟的角度出发,探讨C16TAB分子数对GO最大底面间距的影响,揭示C16TAB在GO层间的排列方式,并结合实验数据进行验证,为季铵盐插层GO对产物结构的影响提供理论依据和借鉴。
1 模拟计算与实验制备
1.1 模型建立与模拟方法
1.1.1 模型建立
在GO结构的实验和理论计算研究基础上[1-4,19],GO结构以石墨单胞为基础原胞,构建12× 12×1的超晶胞,其晶胞大小为2.952 nm×2.952 nm× 0.845 nm[13],-OH与C-O-C异侧邻位均匀分布在碳原子片层两侧,如图1所示。将不同个数的C16TAB分子插入到GO层间,从而构建出C16TAB/GO插层复合物的结构模型,如图2所示。

图1 GO的结构模型Fig.1 Structural model of GO
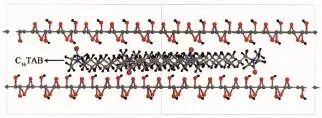
图2 C16TAB/GO插层复合物的结构模型Fig.2 Structural model of C16TAB/GO intercalation compounds
1.1.2 模拟方法
采用基于分子力学的Forcite软件包对相关结构进行结构弛豫与能量优化。分子力学过程采用Smart Minimizer法,UFF(Universal Force Field)力场[20],超软赝势(Ultra-fine)收敛,各项收敛阀值:Energy为8.373 6×10-5kJ·mol-1,Force为4.186 8× 10-2kJ·mol-1·nm-1,Stress为0.001 GPa,Displacement为1.0 fm。迭代步数为10万步,长程静电作用采用Ewald求和法[21],范德华力的截断半径为0.9 nm[21]。
1.1.3 模拟结果
图3(c)为GO结构模型优化后的XRD图。可以看出,优化后的GO结构模型在2θ为10.42°处出现了(001)面网的特征衍射峰,对应的晶面间距为d001= 0.849 nm,这与文献[13]中的实验结果0.845 nm吻合,说明本文的GO结构模型具有合理性。

图3 (a)石墨原样(G)、(b)氧化石墨(GO)样品及(c)GO结构模型的XRD图Fig.3 XRD patterns of original graphite(a),sample(b) and structural model(c)of graphite oxide
图4 (a)为C16TAB/GO插层复合物优化后的层间距与C16TAB分子数的关系图。可以看出,随着C16TAB分子数的增加,C16TAB/GO插层复合物优化后的层间距呈阶梯状逐渐增大,这与Hackett等[14]研究季铵盐改性层状硅酸盐的层间距呈阶梯型变化趋势一致,达到插层饱和值时[13],插层的C16TAB分子数达到28个,形成的阶梯数为5个。当C16TAB分子数分别为1~6、8~12、15~17、19~22和26~28时,层间距分别达到1.56、1.98、2.33、2.76和3.40 nm,分别处于图4(a)中第1~5个阶梯平面。
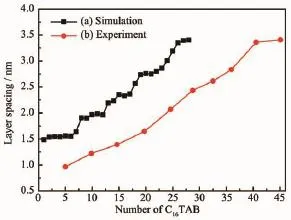
图4 C16TAB/GO插层复合物层间距模拟值、实验值与C16TAB分子数的关系Fig.4 Relationship between the simulation,experimental values about layer spacing and the number of C16TAB molecules in C16TAB/GO intercalation compounds
1.2 实验原料与实验步骤
1.2.1 实验原料
天然鳞片石墨(graphite,G),含碳量90%~99.9%,粒径小于75 μm,青岛申墅石墨制品厂;高锰酸钾(KMnO4)、浓硫酸(H2SO4)、十六烷基三甲基溴化铵(C16TAB)均为分析纯,成都市科龙化工试剂厂;5%的H2O2溶液;5%的HCl溶液;0.05 mol·L-1的NaOH溶液。
1.2.2 实验仪器
电子天平(JT2003,余姚市金诺天平仪器有限公司),超纯水系统(UPT-Ⅱ-10T,成都超纯科技有限公司),恒温水浴锅(DF-101,河南予华仪器有限公司),磁力搅拌器(78HW-1,杭州仪器电机有限公司),恒温振荡器(SHY-100A,长沙湘仪离心机仪器有限公司),恒温干燥箱(202-1,上海圣欣科学仪器有限公司)。
1.2.3 实验步骤
(1)GO的制备:采用文献[13,22-23]中改进的Hummer法制备氧化石墨样品,标记为GO。
(2)C16TAB/GO插层复合物的制备:称取0.2 g GO样品加入到盛有50 mL NaOH溶液(0.05 mol·L-1)的烧杯中,在磁力搅拌器上搅拌均匀,得到GO悬浮液;分别称取不同质量的C16TAB置于烧杯中,加入50 mL的蒸馏水,搅拌使其充分溶解;将溶解后的C16TAB溶液倒入装有GO悬浮液的烧杯中,在恒温振荡器中振荡1 h(20℃);过滤并用去离子水洗涤,洗去未反应的C16TAB和残存的NaOH,用AgNO3溶液(0.1 mol·L-1)检验,直至不出现沉淀物为止,即无Br-;将样品干燥24 h(在60℃烘箱中),获得C16TAB/ GO插层复合物样品,分别标记为GO-16-n(n= 5,10,15,20,25,30,35,40,50,60,70,80)。
1.2.4 实验结果
利用元素分析仪对C16TAB/GO插层复合物中N元素的含量进行了测定,并计算得到实际插入GO层间的C16TAB质量,再根据下式便可以求得插入GO晶胞中的C16TAB分子数。
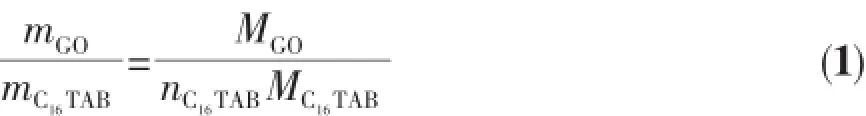
式中:mGO为氧化石墨的质量,且mGO=0.2 g;MGO为氧化石墨结构模型的相对分子质量,GO结构模型中含C原子576个,O原子288个,H原子144个,即MGO=11 664;mC16TAB为插层C16TAB的质量;MC16TAB为C16TAB的相对分子质量,即MC16TAB=364;nC16TAB为插层的C16TAB分子数。
图3(a)、(b)分别为石墨原样(G)和氧化石墨(GO)样品的XRD图。可以看出,GO样品仅在2θ为10.43°处出现了(001)面网的特征衍射峰,对应的层间距为d001=0.848 nm,峰形较为尖锐,且石墨的特征衍射峰消失(d001=0.335 nm),表明GO的氧化程度较好,层堆叠有序度较高,这与图3(c)的模拟结果一致,进一步说明GO结构模型具有合理性。
图5为不同C16TAB分子数插层GO所得C16TAB/GO插层复合物样品的XRD图。从图中可知,C16TAB/GO插层复合物样品的层间距随C16TAB分子数的增加而逐渐增大。当实验中所用C16TAB分子数分别为5、10、15、20、25、30、35、40时,插层复合物样品的层间距随C16TAB分子数的增加逐渐增大,继续增加C16TAB分子数至50、60、70、80时,插层复合物样品的层间距变化较小,保持在3.40 nm左右,表明GO已达到插层饱和状态,这与文献[7,13]中的实验结果一致。因此,C16TAB/GO插层复合物层间距的变化与C16TAB分子数有关。
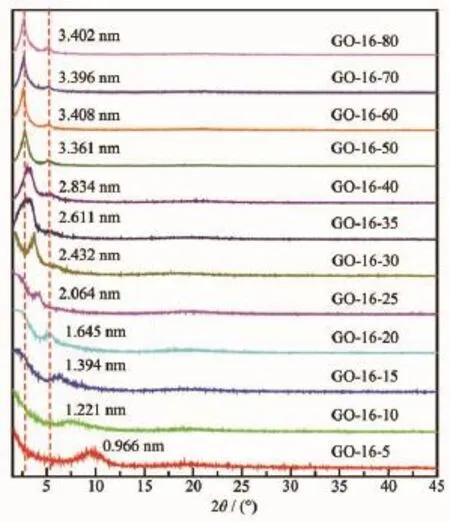
图5 系列C16TAB/GO插层复合物样品的XRD图Fig.5 XRD patterns of series of C16TAB/GO intercalation compound samples
当C16TAB分子数较少时,插层复合物样品的特征衍射峰弥散,比GO样品的特征衍射峰明显宽泛,表明C16TAB分子数较少时,通过离子键插入GO层间,且排列不均匀,即C16TAB在GO层间的排列有序度较差,从而使插层复合物样品的层堆叠有序度较低[7]。当C16TAB分子数逐渐增多时,插层复合物样品的特征衍射峰逐渐锐化,且出现明显的二级衍射峰,说明随C16TAB分子数的增加,C16TAB在GO层间有序化聚集,从而使插层复合物样品的层堆叠有序度增大[24]。
2 结果与讨论
2.1 C16TAB分子数对C16TAB/GO插层复合物层间距的影响
图4为C16TAB/GO插层复合物层间距随C16TAB分子数变化的模拟值(dSim.)与实验值(dExp.)的对比图。从图中可知,C16TAB/GO插层复合物的dSim.和dExp.都随C16TAB分子数的增加而增大,其整体趋势基本一致。不同的是,实验中所用C16TAB分子数呈间隔性增加(不连续),从而使插层复合物的dExp.出现逐渐增加的趋势;而模拟中采用C16TAB分子数增加的幅度为1(连续),插层复合物的dSim.呈现阶梯型增加,分析认为,C16TAB在GO层间某一层排列饱和时才趋于分布下一层,即分层排列,从而导致插层复合物的dSim.在C16TAB分子数区间分别为1~6、8~12、15~17、19~22、26~28时变化很小,即出现阶梯平面。
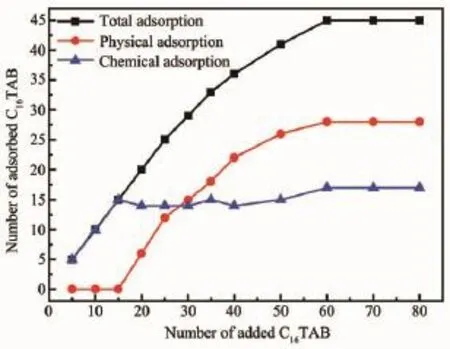
图6 C16TAB分子物理吸附、化学吸附与加入分子数的关系Fig.6 Relationship between the number of physical, chemical adsorption and added C16TAB molecules
根据文献[13,24-26]可知,C16TAB插层GO的过程分为化学吸附(离子吸附)和物理吸附(分子吸附)。图6为GO中C16TAB分子总吸附、化学吸附和物理吸附分子数的关系图,可以看出,当C16TAB加入分子数较少时,C16TAB优先占据GO碳原子层上的化学活性点,并以化学吸附的方式与GO的碳原子层结合,此时C16TAB以化学吸附为主,而GO碳原子层上发生化学吸附的活性点数量是有限的,C16TAB的化学吸附分子数随着C16TAB加入分子数的增加逐渐增加,最后达到饱和吸附,吸附曲线会出现一个平台,这与图6中的化学吸附曲线一致,化学饱和吸附分子数约为17个;C16TAB/GO插层复合物中C16TAB的总吸附和物理吸附分子数随C16TAB加入分子数的增加而逐渐增加,插层饱和时趋于稳定,二者变化趋势基本同步,这表明总吸附分子数的增加主要是物理吸附产生的。
分析图4可知,当dExp.=dSim.,nExp.>nSim.,这主要与C16TAB分子的吸附方式有关,C16TAB分子并没有全部插入GO层间,在化学吸附阶段存在表面吸附和边缘吸附的现象产生,从而反映出实验中使用较多的C16TAB分子数才能达到与模拟一样的d001值。另外,由公式(1)可知,还与GO结构模型的相对分子质量(MGO)有关,本文的GO结构模型只含有-OH、C-O-C两种含氧官能团,且含量均一、分布均匀,没有考虑边缘-COOH、-C=O等含氧官能团的影响,从而减少了化学吸附的分子数;而实验中因实验方法、制备条件的不同,制备的GO结构中含氧官能团的种类、含量以及碳层平面的缺陷程度差异较大[23,27-28]也有一定的影响。
2.2 C16TAB在GO层间的排列模式
C16TAB分子由碳(C)、氢(H)、氮(N)、溴(Br)4种原子组成,根据C16TAB分子的结构式及键长、键角和扭角的数值,并结合各原子的范德华半径,就可以勾勒出C16TAB的形状[29]。由C16TAB的结构模型[30]可知,当“锯齿状”碳链所在平面与基底平面垂直时,亲水端头部和疏水端尾部的截面直径分别为0.51和0.46 nm;当“锯齿状”碳链所在平面与基底平面平行时,亲水端头部和疏水端尾部的截面直径分别为0.67和0.41 nm,对于单碳链呈“锯齿状”排列的C16TAB分子,其链长L为2.53 nm。
根据文献[13,31-33]中季铵盐排列方式的实验研究,C16TAB在GO层间的有效层间域高度(ΔdExp.)等于实测的C16TAB/GO插层复合物的层间距(dExp.)减去所用GO的晶面间距(0.848 nm),即ΔdExp.=dExp.-0.848 nm。结合C16TAB分子的结构及链长(2.53 nm),利用公式θ=arcsin(Δd/L)[34]可以得出C16TAB分子在GO层间的排列方式及C16TAB分子与GO结构层之间的倾角θ(表1)。
由图5可知,当实验中所用C16TAB分子数为5、10、15时,ΔdExp.分别为0.118、0.373、0.546 nm (<0.570 nm),可以判断,此时C16TAB应是单层平躺分布在GO层间[7,13,35];当C16TAB分子数为20、25时,ΔdExp.分别为0.797、1.216 nm,此时C16TAB在GO层间应是双层平躺[7,13,36]或单层倾斜[31,33],倾斜角θ约为18.4°和28.7°;当C16TAB分子数为30、35、40时,ΔdExp.分别为1.584、1.763、1.986 nm,此时C16TAB在GO层间应是三、四层平躺或单层倾斜[7,13,31,33],倾斜角θ约为38.8°、44.2°和51.7°;当C16TAB分子数为50、60、70、80时,ΔdExp.分别为2.513、2.560、2.548、2.554 nm,保持在2.55 nm左右,此时C16TAB在GO层间应是五层平躺或单层直立[13]。
根据上述C16TAB在GO层间排列方式的实验推导,并结合前面的分析结果,针对dSim.的5个阶梯平面,分别建立不同排列方式(平躺、倾斜、直立、混合)的结构模型(C16TAB分子数选取6、12、16、22、28),分为固定d值和不固定d值两类对其进行结构优化处理,从能量(表2、表3)和结构(图7)的角度探讨C16TAB分子在GO层间最优的排列方式。
表2为C16TAB在GO层间不同排列方式优化后的能量表(固定d值)。可以看出,C16TAB在GO层间分层平躺排列时的总能量最低,根据插入分子数的不同,C16TAB在GO层间分别可以形成1~5层平躺排列,而单层倾斜、单层直立和混合排列时的总能量均比对应的平躺排列高,说明C16TAB在GO层间更倾向于平躺排列,此时插层复合物的结构比对应的倾斜、直立和混合时更加稳定。
表3为C16TAB在GO层间不同排列方式优化后的能量及层间距d001值(不固定d值)。可以看出,C16TAB在GO层间也是分层平躺排列时的总能量最低,且层间距d001与dSim.一致,即与图4中的层间距一致,而单层倾斜、单层直立和混合排列时的总能量均比对应的平躺排列高,且优化后的层间距均增大很多,即层间距均超出了相应C16TAB分子数插层时应有的层间距值,说明C16TAB在GO层间单层倾斜、单层直立和混合排列的可能性远小于平躺排列。

表1 C16TAB分子在GO层间的排列方式(实验)Table1 Arrangement modes of C16TAB molecules in the interlayer of GO(experiment)

表2 C16TAB在GO层间不同排列方式优化后的能量(固定d值)Table2 Energy of different arrangement modes of C16TAB molecules in the interlayer of GO after minimization(fixed value of d)

表3 C16TAB在GO层间不同排列方式优化后的能量及d001值(不固定d值)Table3 Energy and d001of different arrangement modes of C16TAB molecules in the interlayer of GO after minimization(not fixed value of d)
当C16TAB分子插入量一定时,C16TAB也可能存在不同的分层排列,在此以C16TAB分子数为22时(GO-16-22)为例,建立1~4层平躺排列时插层复合物的结构模型(固定c值为2.76 nm,以排除碳原子结构层面移动的影响),并对其进行结构优化,优化前后插层复合物的结构模型如图7所示。
图7为C16TAB/GO插层复合物优化前后的结构模型。可以看出,在结构优化的过程中,为使复合物结构体系达到最稳定,C16TAB在GO层间做分子运动,寻找能量最低的稳定位置,对比优化前后的结构可知,GO层间的C16TAB由优化前的初始状态全部转变为4层平躺排列;且从结构能量角度(表4)比较可知,C16TAB在GO层间1~4层平躺排列优化后的体系结构能量有E1>E2>E3>E4,即4层平躺排列能量最低、结构最稳定,表明C16TAB在GO层间的最佳位置趋向于分层平躺排列,与前面分析结果一致。另外,优化前碳原子结构层面较平整,优化后碳原子结构层面出现不同程度的弯曲和变形,这是由于在结构优化的过程中c轴被固定,碳原子结构层不能通过上下左右移动来降低体系的能量,只能通过结构层面上碳网的弯曲与变形来使体系更加稳定,从而出现碳原子结构层面的弯曲及变形。

图7 C16TAB/GO插层复合物优化前后的结构模型(C16TAB分子数为22)Fig.7 Structural models of C16TAB/GO intercalation compounds before and after minimization (Number of C16TAB molecules is 22)
表4 C16TAB/GO插层复合物优化前后的能量(C16TAB分子数为22)Table4 Energy of C16TAB/GO intercalation compounds before and after minimization(Number of C16TAB molecules is 22)
综合上述分析,当dSim.=1.56 nm时,单层平躺的总能量比单层倾斜和混合排列的低,且层间距与dSim.一致,此时C16TAB在GO层间的最优排列为单层平躺[13-15,35-36];当dSim.=1.98 nm时,二层平躺的总能量比单层倾斜和混合排列的低,且层间距与dSim.吻合,此时的最优排列为二层平躺[13-15,35-36];当dSim.=2.33 nm时,C16TAB插入分子数较少,且ΔdSim.<L(2.53 nm),C16TAB分子不可能在GO层间单层直立,由单层直立结构模型(假设)优化后的总能量最高可知,此时插层复合物的结构并不稳定,即此时单层直立排列的结构存在性极小,进一步说明能量优化的合理性,由三层平躺的总能量比单层倾斜、单层直立和混合排列的低以及层间距与dSim.吻合可知,此时的最优排列为三层平躺,这与文献[14-15]中烷基链在硅酸盐层间呈现假三层分布类似;当dSim.=2.76 nm时,四层平躺的总能量比单层倾斜、单层直立和混合排列的低,且层间距与dSim.一致,此时的最优排列为四层平躺,这与文献[15]中烷基链在蒙脱石层间呈现假四层分布类似;当dSim.=3.40 nm时,五层平躺的总能量比单层倾斜、单层直立和混合排列的低,且层间距与dSim.一致,此时的最优排列为五层平躺。因此,根据插入分子数的不同,C16TAB在GO层间的最优排列为1~5层平躺排列。
3 结论
(1)GO结构模型优化后的层间距为0.849 nm,模拟结果与实验数据吻合,模拟结构具有合理性。
(2)C16TAB/GO插层复合物模拟计算的层间距随C16TAB分子数的增加呈5个阶梯状逐渐增大,层间距的实验值随着C16TAB分子数的增加逐渐增大,插层饱和的层间距均为3.40 nm,实验结果与模拟结果能够很好地吻合。
(3)C16TAB在GO层间的排列方式与C16TAB的分子数有关,模拟结果显示C16TAB在GO层间的最优排列方式为分层平躺,根据插入C16TAB分子数的不同呈现1~5层平躺排列。
[1]Nakajima T,Mabuchi A,Hagiwara R.Carbon,1988,26(3): 357-361
[2]He H,Klinowski J,Forster M,et al.Chem.Phys.Lett., 1998,287(1):53-56
[3]Lerf A,He H,Forster M,et al.J.Phys.Chem.B,1998,102 (23):4477-4482
[4]Boukhvalov D W,Katsnelson M I.J.Am.Chem.Soc.,2008, 130(32):10697-10701
[5]YANG Jian-Guo(杨建国),NIU Wen-Xin(牛文新),LI Jian-She(李建设),et al.Polym.Mater.Sci.Eng.(高分子材料科学与工程),2005,21(5):55-58
[6]Mermoux M,Chabre Y,Rousseau A.Carbon,1991,29(3): 469-474
[7]Matsuo Y,Niwa T,Sugie Y.Carbon,1999,37(6):897-901
[8]Xu S,Boyd S A.Environ.Sci.Technol.,1995,29(2):312-320 [9]Williams D S,Thomas R K,Castro M A,et al.J.Colloid Interface Sci.,2003,267(2):265-271
[10]HAN Zhi-Dong(韩志东),WANG Jian-Qi(王建祺).Chinese J.Inorg.Chem.(无机化学学报),2003,5(19):459-461
[11]Liu Z H,Wang Z M,Yang X J.Langmuir,2002,18(12):4926-4932
[12]Matsuo Y,Miyabe T,Fukutsuka T,et al.Carbon,2007,45 (5):1005-1012
[13]LIN Shun-Jia(林舜嘉),SUN Hong-Juan(孙红娟),PENG Tong-Jiang(彭同江),et al.Chinese J.Inorg.Chem.(无机化学学报),2013,29(11):2333-2338
[14]Hackett E,Manias E,Giannelis E P.J.Chem.Phys.,1998, 108(17):7410-7415
[15]Zeng Q H,Yu A B,Lu G Q,et al.Chem.Mater.,2003,15 (25):4732-4738
[16]Miroslav P,Pavla C,Dagmar M,et al.J.Colloid Interface Sci.,2001,236(1):127-131
[17]Miroslav P,Pavla C,Dagmar M,et al.J.Colloid Interface Sci.,2002,245(1):126-132
[18]FU Yi-Zheng(付一政),LIAO Li-Qiong(廖黎琼),LIANG Xiao-Yan(梁晓艳),etal.Polym.Mater.Sci.Eng.(高分子材料科学与工程),2013,29(7):175-178
[19]Liu B,Sun H J,Peng T J,et al.J.Mol.Model.,2012:1-6
[20]Rappé A K,Casewit C J,Colwell K S,et al.J.Am.Chem. Soc.,1992,114(25):10024-10035
[21]Frenkel D,Smit B,Translated by WANG Wen-Chuan(汪文川),ZHOU Jian(周健),CAO Da-Peng(曹大鹏).Understanding Molecular Simulation-From Algorithms to Applications (分子模拟-从算法到应用).Beijing:Chemical Industry Press,2002:329-338
[22]FU Ling(傅玲),LIU Hong-Bo(刘洪波),ZOU Yan-Hong(邹艳红),et al.Carbon(炭素),2005(4):10-14
[23]HUANG Qiao(黄桥),SUN Hong-Juan(孙红娟),YANG Yong-Hui(杨勇辉).Chinese J.Inorg.Chem.(无机化学学报),2011,27(9):1721-1726
[24]DING Yun-Sheng(丁运生),WANG Seng-Shan(王僧山), ZHA Min(查敏).Acta Phys.-Chim.Sin.(物理化学学报), 2006,22(5):548-551
[25]Le P L,Duchet J,Sautereau H,et al.Macromol.Symp., 2003,194(1):155-160
[26]LIN Bao-Hui(林宝辉),GAO Mang Mang-Lai(高芒来).Acta Phys.-Chim.Sin.(物理化学学报),2005,21(7):808-812
[27]LI Lin-Jiang(李林江),HU Dong-Hu(胡栋虎),JI Ling-Li(季伶俐),et al.J.Funct.Mater.(功能材料),2011,42(B02):168-172
[28]ZOU Yan-Hong(邹艳红),LIU Hong-Bo(刘洪波),FU Ling (傅玲),et al.Journal of the Chinese Ceramic Society(硅酸盐学报),2006,34(3):318-323
[29]ZHOU Gong-Du(周公度).Structural Chemistry of Inorganic (无机结构化学).Beijing:Science Press,1984:118-119
[30]He H P,Frost R L,Bostrom T,et al.Appl.Clay Sci.,2006, 31(3):262-271
[31]Zhu J X,He H P,Guo J G,et al.Chin.Sci.Bull.,2003,48 (4):368-372
[32]Williams D S,Thomas R K.J.Colloid Interface Sci.,2002, 255:303-311
[33]Slade P G,Gates W P.Appl.Clay Sci.,2004,25(1):93-101
[34]CHEN De-Fang(陈德芳),WANG Zhong(王重).J.Xi′an Jiaotong University(西安交通大学学报),2000,34(8):92-95
[35]Vaia R A,Teukolsky R K,Giannelis E P.Chem.Mater., 1994,6(7):1017-1022
[36]Vahedi F A,Guggenheim S.Clays Clay Miner.,1997,45(6): 859-866
Molecular Simulation of Structure of Cetyl Trimethyl Ammonium Bromide Intercalated Graphite Oxide
ZHAO Er-Zheng1PENG Tong-Jiang*,1,2SUN Hong-Juan1LIU Bo1JI Guang-Fu3
(1Key Laboratory of Solid Waste Treatment and Resource Recycle,Southwest University of Science and Technology,Ministry of Education,Mianyang,Sichuan 621010,China)
(2Center of Analysis and Test,Southwest University of Science and Technology,Mianyang,Sichuan 621010,China)
(3National Key Laboratory of Shock Wave and Detonation Physics,Institute of Fluid Physics, China Academy of Engineering Physics,Mianyang,Sichuan 621900,China)
The structure changes of C16TAB/GO intercalation compounds about the number of cetyl trimethyl ammonium bromide(C16TAB)molecules were investigated by molecular simulation method.The arrangement modes of C16TAB molecules in the interlayer of GO were discussed,and the simulation results were verified by the experimental data.The simulation results show that the layer spacing of the structural model of GO is 0.849 nm;the layer spacing of C16TAB/GO intercalation compounds increases gradually by five ladderlike style with the increase of the number of C16TAB molecules.The layer spacing of each ladder are 1.56,1.98,2.33,2.76 and 3.40 nm,and the number of C16TAB molecules is up to 28 when the intercalation is saturated.The experimental results show that the layer spacing of C16TAB/GO intercalation compounds increases gradually with the increase of the number of C16TAB molecules and the saturation value is 3.40 nm,so the experimental results are in good agreement with simulation results.The possible arrangement modes of C16TAB molecules in the interlayer of GO are 1~5 layers lateral arrangement or lateral monolayer,paraffin-type monolayer and vertical monolayer,and theoptimal arrangement modes of C16TAB molecules in the interlayer of GO are 1~5 layers lateral arrangement according to the result of energy and structure.
cetyl trimethyl ammonium bromide;graphite oxide;molecular simulation;layer spacing;arrangement mode
O641
A
1001-4861(2015)03-0485-08
10.11862/CJIC.2015.081
2014-08-27。收修改稿日期:2014-12-12。
国家自然科学基金(No.41272051)和西南科技大学博士基金(No.11ZX7135)资助项目。
*通讯联系人。E-mail:tjpeng@swust.edu.cn,Tel:+86-816-2419016;会员登记号:S130010901S。
猜你喜欢
发明与创新(2022年34期)2022-12-01
分子催化(2022年1期)2022-11-02
特区文学·诗(2021年6期)2021-12-22
无机材料学报(2020年1期)2020-02-10
电子制作(2019年15期)2019-08-27
表面工程与再制造(2019年6期)2019-08-24
中国生殖健康(2019年8期)2019-01-07
青春(2017年5期)2017-05-22
浙江大学学报(工学版)(2016年2期)2016-06-05
中国塑料(2016年11期)2016-04-16
