
伊潘立酮的合成
2015-10-21 18:30钟霞陈年根张万科张俊清
科技与企业 2015年19期
钟霞 陈年根 张万科 张俊清
引言
目的:研究用于治疗精神分裂的药物伊潘立酮的制备方法。方法:以3-甲氧基-4-羟基苯乙酮和3-碘-1-丙醇为起始原料,经亲核取代反应、磺酸酯化和N-烷基化三步制备得到目标产物。结果:H-NMR和文献报道的一致,总收率为45.8% (以香草酮计)。
结论:本制备方法克服了文献所报道的制备工艺的缺点,与现有技术相比,本发明后处理更简单,更适合工业化生产。
伊潘立酮(iloperidone, 1),化学名为4-[3-[4-(6-氟-1, 2-苯并异恶唑-3-基)-1-哌啶基]丙氧基]-3-甲氧基苯乙酮,是一种新型非典型抗精神分裂药物。与目前常用的抗精神病药物比较,短期、长期的安全试验结果显示,伊潘立酮的副作用更少,不会诱导患者发生糖尿病,患者锥体外系症状也较少。
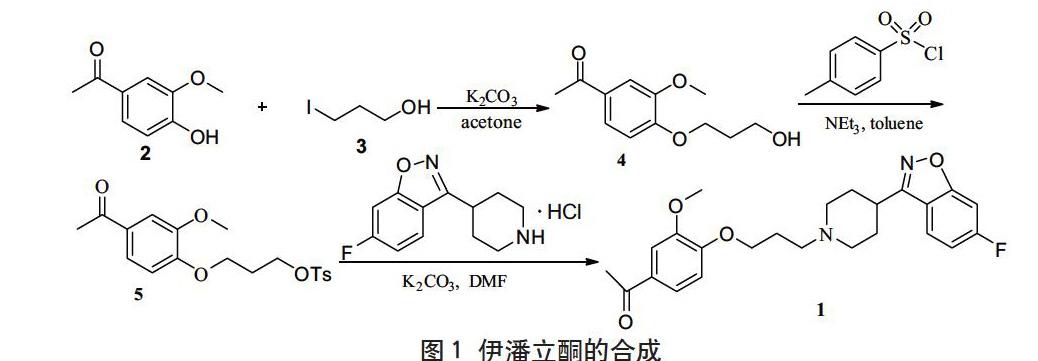
本文中所采用的伊潘立酮的合成路线见图1。
图1 伊潘立酮的合成
Figure 1 The synthetic route of iloperidone
1、实验部分
1.1主要仪器与试剂
X4型显微熔点测定仪(温度未经校正)、Bruker-AV-400型核磁共振仪(溶剂CDCl3,内标TMS)。所用试剂为工业品或市售分析纯。
1.2实验步骤
1.2.1 3-甲氧基-4-(3-羟基丙氧基)苯乙酮(4)的合成
于2 L三口烧瓶中加入香草酮(2,166.2g,1mol),丙酮(800ml),搅拌使溶,再加入无水K2CO3(150g,1.09mol)。滴加3-碘-1-丙醇(3,195.3g,1.05mol),滴畢回流反应8h。冷却至室温,减压抽滤,滤饼用丙酮(100ml)洗涤,减压回收溶剂。残留液中加入石油醚(300ml),室温搅拌析晶4 h。抽滤,滤饼用石油醚(50ml*2)洗涤,40℃鼓风干燥4h,得微黄色固体(195.8g)。
1.2.2 3-(2-甲氧基-4-乙酰基苯氧基)-1-丙醇对甲苯磺酸酯(5)的合成将4(112g,0.5mol)、甲苯(600ml)、三乙胺(58.1g,0.575 mol)加入到2L三口烧瓶中,搅拌溶解,冰水浴下滴加对甲苯磺酰氯(104.8g,55mol),控制温度0~10℃。滴毕,维持温度搅拌反应10h。加入蒸馏水(100ml),有机相分别用饱和NaHCO3水溶液(50ml)、蒸馏水洗涤(50ml),无水硫酸钠干燥。减压蒸除约2/3溶剂,再加入正己烷(400ml)。冷却至室温。减压过滤,滤饼用石油醚(30ml*2)洗涤,45℃鼓风干燥4h,得白色固体(147g)。
1.2.3 伊潘立酮(1)的合成。于2L反应釜中,加入6-氟-3-(4-哌啶基)-1,2苯并异噁唑盐酸盐(77g,0.3mol),5(113.5g,0.3mol)、N, N-二甲基甲酰胺(500ml)、K2CO3(82.8g,0.6mol)、KI(2g),搅拌混匀,加热至80~90℃反应8h,冷却至室温,过滤,滤液减压浓缩,残留液加入二氯甲烷(500ml)、蒸馏水洗涤(50ml)。分液,无水硫酸钠干燥,减压回收溶剂,得淡黄色固体,丙酮-乙酸乙酯重结晶得白色固体(86.4g)。
2、结果
2.2.1 3-甲氧基-4-(3-羟基丙氧基)苯乙酮(4)的1H-NMR数据
得微黄色固体(195.8g,收率87.4%),m.p.120~123℃。1H-NMR (400MHz,CDCl3)δ:7.54(d,J=8.5Hz,1H),7.59(s,1H),6.90(d, J=8Hz,1H),4.26(t,2H),3.91(s,3H),3.87(t,2H),2.63(s,1H,OH), 2.56(s,3H),2.12(d,J=8Hz,2H)。
2.2.2 3-(2-甲氧基-4-乙酰基苯氧基)-1-丙醇对甲苯磺酸酯(5)的1H-NMR数据得白色固体(147g,收率77.7%),m.p.94~97℃。1H-NMR(400MHz,CDCl3)δ:7.76(d,J=8Hz,2H),7.51(d,J=8.5Hz,1H),7.49(s,1H),7.25(d,J=8Hz,2H),6.80(d,J=8Hz,1H),4.28(t,2H),4.07(t,2H),3.99(s,3H),2.57(s,3H),2.37(s,3H),2.20(d,J=8Hz, 2H)。
2.2.3 伊潘立酮(1)的1H-NMR数据
得白色固体(86.4g,收率67.5%),m.p.121~123℃(文献[2], m.p.121~123℃)。1H-NMR(400 MHz,CDCl3)δ:7.73(d,J=8Hz, 1H),7.50(d,J=8.5Hz,1H),7.47(s,1H),7.46(s,1H),7.22(d, J=8Hz,2H),6.78(d,J=8Hz,1H),4.23(t,2H),3.92(s,3H) 3.07~3.09(m,3H),2.57(t,2H),2.51(s,3H),2..20(m,2H), 1.72~2.10(m,6H)。
3、讨论
文献报道的合成路线主要有三种:路线一先制备(2,4-二氟苯基)-哌啶-4-基甲酮肟盐酸盐与3-甲氧基-4-(3-氯-1-丙基)苯乙酮,二者再发生N-烃化缩合反应,关环得1。
路线二以3-甲氧基-4-羟基苯乙酮为起始物料,与1-溴-3-氯丙烷反应得到3-甲氧基-4-(3-氯-1-丙基)苯乙酮,再与6-氟-3-(4-哌啶基)-1, 2-苯并异恶唑盐酸盐,在N, N-二甲基甲酰胺和乙腈的混合溶剂中,K2CO3催化下,反应得1。路线一环合作为最后一步反应,能提高反应的区域选择性,减少副产物。但是环合反应收率较低,增加合成的成本。
同时在强碱条件下,不利于产物的稳定和最终产品的质量控制;路线二采用了聚合式的合成路线,极大提高原子经济性。但是在制备3-甲氧基-4-(3-氯-1-丙基)苯乙酮时,采用了具有区位选择性的取代反应,该反应区位选择性不够强,需用1-溴-3-氯丙烷较大的过量才能实现较好选择性。反应副产物多,分离难度增加,增加经济成本。上述两种合成路线均存在一定的问题,产品分离困难。高晓婷等用3-甲氧基-4-羟基苯乙酮和3-氯-1-丙醇反应,再磺酸酯化,可避免路线二的副反应,但没有提到终产物伊潘立酮的合成。我们在路线三的基础上,以活性更强的3-碘-1-丙醇(3)代替3-氯-1-丙醇,和3-甲氧基-4-羟基苯乙酮(2)反应,以增加区位的选择性,所得的3-甲氧基-4-(3-羟基丙氧基)苯乙酮(4)再在对甲苯磺酰氯的作用下生成磺酸酯(5),其和6-氟-3-(4-哌啶基)-1, 2-苯并异恶唑盐酸盐缩合得。本路线副反应少,反应条件温和,后处理简单,这对于产品的工业化具有十分重要的意义。
(作者单位:海南医学院药学院)
作者简介
钟霞(1978-),女,甘肃兰州人,博士,副教授,研究方向:药物合成。
注:*海南省自然科学基金(No.814287)资助课题
猜你喜欢
学苑创造·B版(2019年11期)2019-12-05
小猕猴智力画刊(2018年3期)2018-06-12
中学课程辅导·教学研究(2017年29期)2018-02-26
小学生导刊(低年级)(2016年8期)2016-09-24
中国医药科学(2016年9期)2016-07-25
数理化学习·高一二版(2009年1期)2009-03-19
中国科技术语(2004年4期)2004-03-18
