
基于纳米通道表面增强拉曼散射光谱分离检测组氨酸对映体的研究
2015-11-16 18:06钟桐生尹志芳柳悦黄杉生
分析化学 2015年11期
钟桐生+尹志芳+柳悦+黄杉生

摘 要 以Al2O3纳米通道膜为基体制备金纳米通道,以场发射扫描电镜、循环伏安、交流阻抗等方法对金纳米通道进行表征。采用EDC-NHS的交联反应,将壳聚糖自组装至金纳米通道孔壁上,形成表面具有手性位点选择性的功能化纳米通道膜,利用纳米通道优异的分离能力手性分离D-,L-组氨酸。考察了纳米通道孔径和溶液的pH值对分离效果的影响。采用银溶胶作为表面增强拉曼(SERS)测试的基底,增强对D-,L-组氨酸的SERS效应,提高检测该物质的选择性和灵敏度。分别在1000和1590 cm处测定L-组氨酸和D-组氨酸。在含200 μL组氨酸、100 μL银溶胶和100 μL 80 mmol/L NaCl 溶液(pH=7.59)中,D-组氨酸和L-组氨酸可得到较好分离,分离度达到4.91。
关键词 金纳米通道; 手性分离; 表面增强拉曼光谱; 组氨酸
1 引 言
手性是生物体系的一个基本特征,很多内源性大分子物质,如酶、载体、受体、血浆蛋白和多糖等都具有手性特征[1]。天然或半合成药物几乎都有手性,但产生不同的药理作用和反应。手性对映体的分离测定,对研究生命科学、药物化学以及人类健康都具有重要的意义[2~4]。氨基酸是组成蛋白质的基本单元,在人体生命活动中起着举足轻重的作用,氨基酸多为外消旋体,D型氨基酸在生理活性和实际用途与L型有很大差别,因此D-,L-氨基酸对映体的分离是生命科学研究的基础内容之一,在蛋白质多肽的研究、有机化学中的不对称合成以及医药、食品、卫生等领域的研究中都具有重要意义。常用的手性对映体的分离方法有化学分离法、结晶拆分法、酶或微生物拆分法、萃取拆分法、化学传感器和色谱等方法[5~9]。此外,膜分离法也被广泛用于对映体的分离[10~12]。
表面增强拉曼光谱(SERS)是表征表面分子吸附行为和分子结构的有力工具, 已成为高灵敏的研究界面效应的技术之一,广泛应用于研究吸附分子在表面的取向及吸附行为、吸附界面表面状态,生物大分子的界面取向及构型、构象和结构分析[13,14]。SERS已成为一个强大的分析工具,当物质分子吸附到粗糙金属上(例如Au, Ag, Cu等),在很低的浓度甚至单分子都能有SERS效应[16~19]。
纳米通道已在生物医药、生命科学领域的分离分析研究中显示出高度的优越性和实用性[19~23]。但是采用纳米通道的方法分离D-,L-组氨酸,且同时利用表面增强拉曼光谱测定被分离的物质鲜有报道。本研究以氧化铝膜为基底制备金纳米通道阵列,壳聚糖在EDC-NHS的交联作用下自组装至金纳米通道孔壁上,形成表面具有手性位点选择性的功能化纳米通道膜,利用纳米通道优异的分离能力手性分离D-,L-组氨酸。采用银溶胶作为表面增强拉曼的基底,增强对D-,L-组氨酸的SERS效应,可对D-组氨酸和L-组氨酸同时检测。本研究为构建纳米通道分离池与SERS检测系统的偶联装置,实现对被分离物质的实时检测、深入理解分离机理打下实验基础,体现了其独特的优越性和广阔的应用前景。
2 实验部分
2.1 仪器与试剂
CHI-760B电化学工作站(上海辰华公司); U形连通池(自制); 旋光仪(Autopol IV/IV, USA); 拉曼光谱仪(RENISHAW,英国); S4800场发射扫描电镜(FESEM, HITACHI,日本)。
孔径为100 nm的氧化铝膜购自Waterman 公司; D-组氨酸、L-组氨酸(TCI);1-(3-二甲氨基丙基)-3-乙基硫二亚胺(EDC, 98.5%)、N-羟基琥珀酰亚胺(NHS, 99%)壳聚糖(CS)及3-巯基丙酸(99%)由Aldrich 公司提供;其余试剂均为分析纯,实验用水为超纯水(18.2 MΩ cm)。
按文献[24]所述方法, 用柠檬酸钠还原法制备银溶胶。取0.0255 g AgNO3溶于150 mL超纯水中,不断搅拌,并将溶液加热至沸腾后,取3 mL 1%柠檬酸钠溶液逐滴缓慢加入其中。在沸腾状态下继续加热溶液10 min,同时不断搅拌,停止加热,自然冷却至室温,得到呈灰色的银溶胶。避光保存。
制备的银溶胶以紫外可见光谱进行表征。所制备的银溶胶的最大吸收峰在418 nm处,这是由于金属粒子表面的等离子共振激发或带间跃迁,金属胶体在紫外可见区有吸收带或吸收区。并且在最大吸收峰之后并没有其它的峰,这说明制得的银溶胶的纳米颗粒粒径分布均匀,没有团聚。因此,所制备的银溶胶适合用作SERS活性基底。
2.2 实验方法
2.2.1 金纳米通道的制备与修饰 以Al2O3纳米通道膜为基体,依文献[25]方法制备Au纳米通道膜。将所制得的Au纳米通道膜浸入1%巯基丙酸溶液中,6 h后用去离子水冲洗若干次,然后浸入(5∶1, V/V)EDC-NHS溶液中, 2 h后用水冲洗干净,将活化的膜浸没在0.4%(w/w)的壳聚糖溶液中(pH=7.4)24 h,通过EDC-NHS交联的壳聚糖自组装在Au纳米通道膜上,用水冲洗干净后备用。实验均在4℃条件下进行。为简便计,分别以Al2O3, Al2O3/Au和Al2O3/Au/CS表示Al2O3纳米通道膜、沉积了金的Al2O3纳米通道膜和修饰了壳聚糖的Al2O3/Au膜。
2.2.2 纳米通道传感装置 采用文献[25]所用的U形池作为色氨酸对映体分离装置,以氧化铝纳米通道膜作为分离载体,将膜置于两U形池进样池、透过池之间,膜的有效透过面积为0.196 cm2。在U 形连通池的进样池中加入104 mol/L D-组氨酸和10
@@ 4 mol/L L-组氨酸各4 mL, 透过池中加入4 mL水和4 mL银胶溶液,间隔一定时间用拉曼光谱仪检测透过池中D-组氨酸和 L-组氨酸的量,作出渗透池中物质的量随时间的关系,所得直线斜率之比(α)定义为两种待测物的分离度。endprint
3 结果与讨论
3.1 金纳米通道和修饰膜的表征
图1为以100 nm孔径Al2O3膜为基底的纳米通道膜, 沉积不同时间得到的金纳米通道膜的FESEM图。由图可见,随着镀金时间的增加,Al2O3膜的孔径不断减小, 表明Au纳米粒子已成功沉积在了基底膜的表面及孔道内。
交流阻抗谱和循环伏安法是考察纳米通道制备过程的有效方法[24]。由Al2O3纳米通道膜在0.1 mol/L K3[Fe(CN)6]溶液中的循环伏安图(图2)可见,金沉积时间延长,电流下降。可能是由于随着金沉积时间延长,纳米通道的孔径变小,电子转移阻力增大所致。图3为纳米通道膜在0.1 mol/L K3[Fe(CN)6]溶液中的交流阻抗谱图。由图3可见,Al2O3纳米通道膜经过金的沉积后,由于通道孔径变小,阻抗变大;而Al2O3/Au膜修饰了CS后,孔径进一步减小,其阻抗也进一步增加,表明纳米通道被成功修饰。
3.2 纳米通道孔径对分离效果影响
纳米通道的孔径可通过金沉积时间控制。考察了不同金沉积时间后得到的金纳米通道膜,经壳聚糖修饰后对手性组氨酸分离的影响(图4)。实验表明,金沉积时间太短,纳米通道孔径太大,D-组氨酸和 L-组氨酸均能通过纳米通道,但金沉积时间太长,则通道孔径太小,甚至使得通道被堵死,以至组氨酸不能通过通道。 在100 nm的裸氧化铝膜上镀金9 h后得到的纳米通道膜对D-,L-组氨酸的分离效果最好。
3.3 溶液的pH值对D,L-组氨酸分离度的影响
分别将适量pH为2.0, 3.5, 5.0, 5.6, 7.6和9.0的D-, L-组氨酸溶液及等体积空白溶液加入进样池中,每间隔1 h后检测池D-组氨酸和L-组氨酸含量。从图5可见在低pH或者高pH下,Al2O3/Au/CS对D-,L-组氨酸的分离度较小; 而在pH=7.59时分离度较大。即在组氨酸等电点(pH=7.59)时的分离度最大,达到α=4.5。
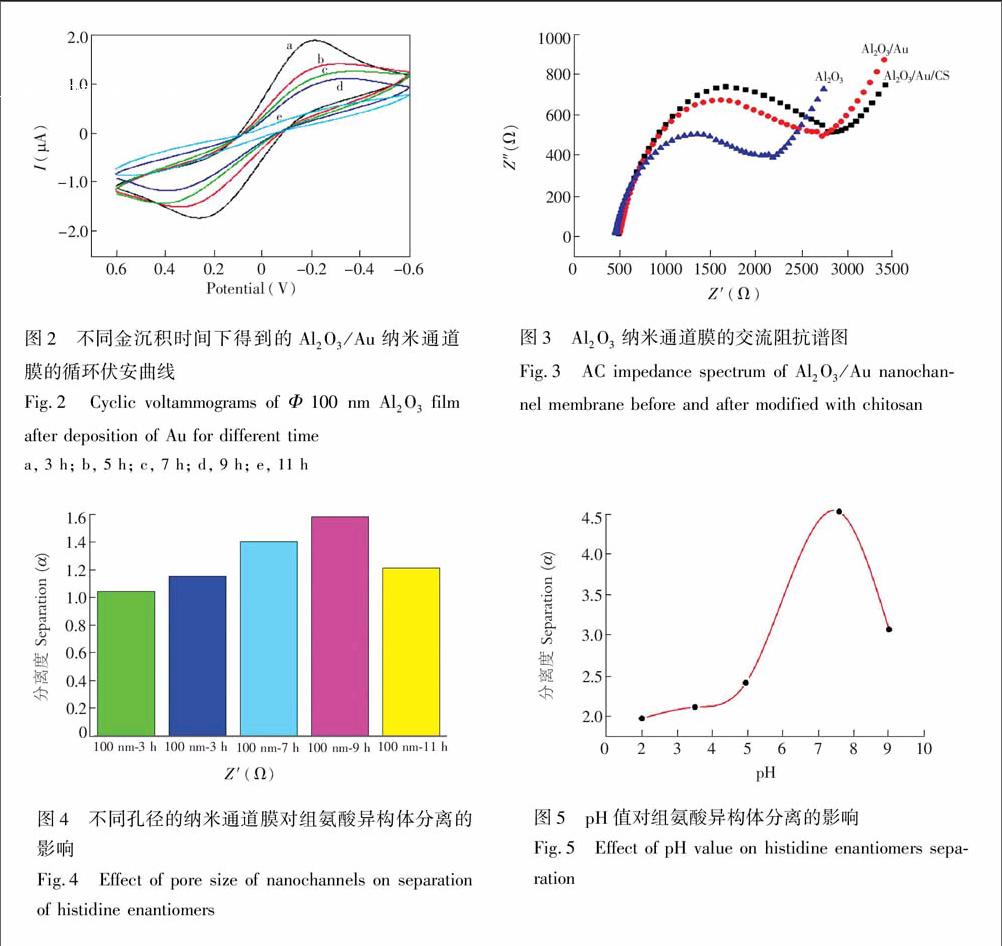
10 mol/L L-组氨酸以及相同浓度的D-,L-组氨酸混合物的SERS光谱。分别取200 μL组氨酸、100 μL银溶胶和100 μL 80 mmol/L NaCl,混合均匀。从图6d可见, 银溶胶起到了SERS活性基底的作用,未加入活性基底时, 组氨酸无法检出。由于D-组氨酸和L-组氨酸的空间位阻不同,与银微粒作用不同,振动频率不同,所以会出现各自的特征峰。L-组氨酸和D-组氨酸分别在1000 cm
1(图6b)和1590 cm
(图6c)处有各自的特征峰; 而在D-,L-组氨酸的混合溶液中, 通过SERS检测, 可以同时看到D-组氨酸和L-组氨酸的特征峰存在(图6a),表明SERS可以同时检测、区分D-组氨酸和L-组氨酸。
3.5 基底团聚时间对SERS检测的影响
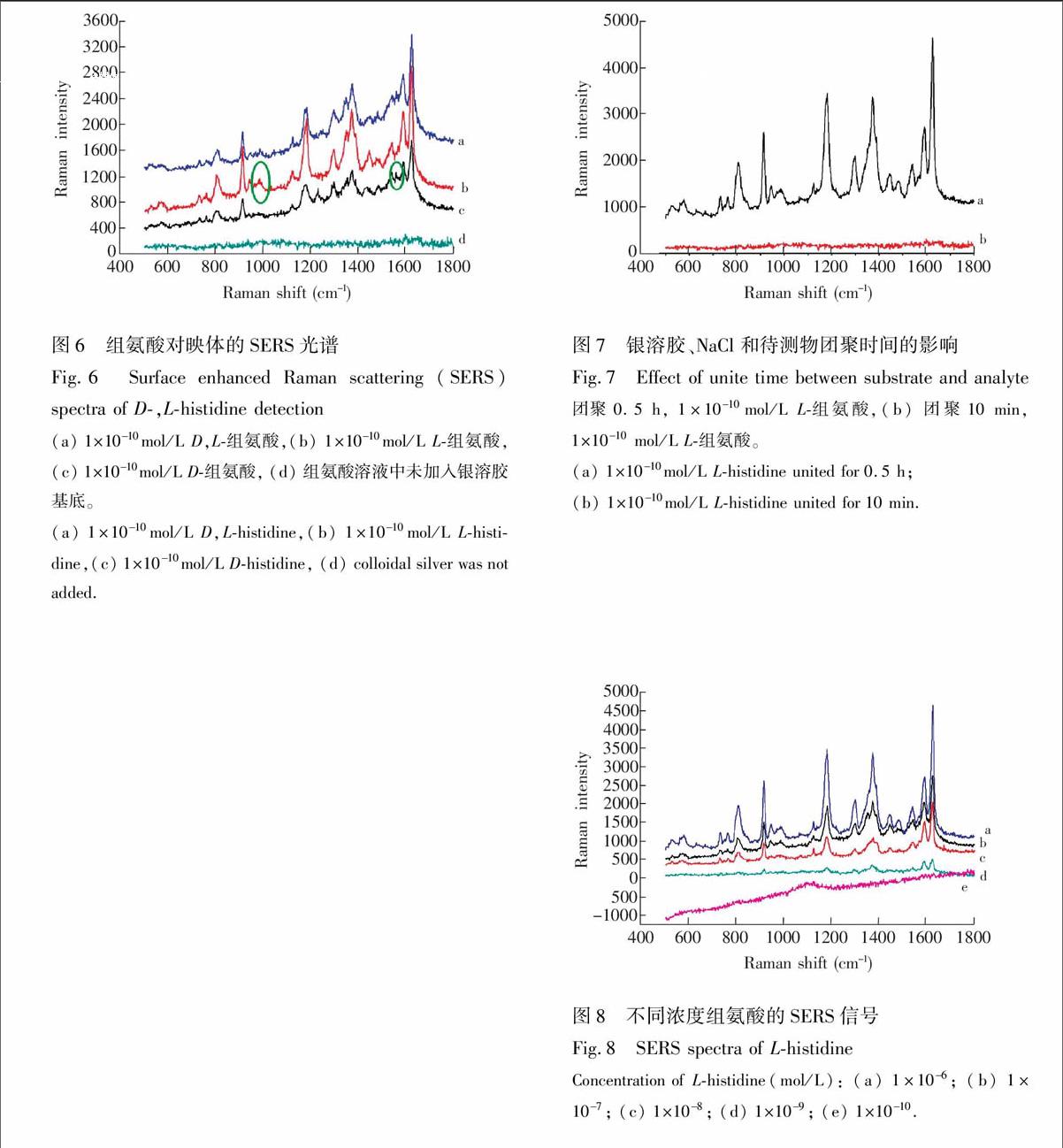
银溶胶和组氨酸混合后的静置时间影响组氨酸的SERS信号。考察了银溶胶、NaCl和待测物团聚时间的影响(图7)。以制备好的银溶胶作为基底,分别取200 μL 1×1010 mol/L组氨酸、100 μL银溶胶和100 μL 80 mmol/L NaCl,混合均匀。结果表明, 团聚时间对分析物检测有很大的影响。团聚时间太短, 分析物与银溶胶未很好地吸附,导致银溶胶没有起到SERS增强的效果,所以没有SERS信号,无法检测出待测物质。实验中,银溶胶和组氨酸混合1 h后再进行SERS信号的检测。
10 mol/L D-组氨酸, (d) 组氨酸溶液中未加入银溶胶基底。
(a) 1×1010mol/L D,L-histidine,(b) 1×1010mol/L L-histidine,(c) 1×0
10mol/L D-histidine, (d) colloidal silver was not added.
团聚0.5 h, 1×1010 mol/L L-组氨酸,(b) 团聚10 min, 1×10
10 mol/L L-组氨酸。
(a) 1×10
10 mol/L L-histidine united for 0.5 h;
(b) 1×10
10 mol/L L-histidine united for 10 min.
3.6 组氨酸检出限的考察
3.7 组氨酸对映体在Al2O3/Au/CS中的迁移
考察了D-,L-组氨酸在修饰前的纳米通道内的迁移量随时间的变化。由图9A可见, Al2O3/Au纳米通道膜对组氨酸对映体没有选择性,二者在金纳米通道内的迁移速率基本上相同,所以不能将两者分离。图9B为D-组氨酸、L-组氨酸通过Al2O3/Au/CS纳米通道膜迁移量随时间的变化。选择在pH为7.59的条件下对1×10
5 mol/L D-,L-组氨酸对映体进行拆分。很显然,D-组氨酸的迁移速率明显大于L-组氨酸,壳聚糖功能化的金纳米通道对D-,L-组氨酸对映体分离度为4.91。这是由于表面具有手性位点选择性的壳聚糖功能化的金纳米通道膜对手性组氨酸具有优异的分离能力。
图9 1×10
5 mol/L组氨酸手性对映体过Al2O3/Au纳米通道膜(A)和过Al2O3/Au/CS纳米通道膜(B)的分离情况
Fig.9 Separation efficiency of histidine enantiomers through Au nanochannels membrane (A) and chitosan functionalized Au nanochannels membrane (B)
4 结 论
以氧化铝为模板,壳聚糖功能化的金纳米通道在最佳条件下对D-,L-组氨酸对映体分离度最大达到4.91。由于SERS检测的检出限低,SERS可以同时检测出D-组氨酸和L-组氨酸,可望与纳米通道进行偶联,在线实时分离检测手性组氨酸对映体,能够大大缩短检测时间。本研究为构建纳米通道分离池与SERS检测系统的偶合装置, 对快速便捷地分离检测手性物质提供了方法,具有潜在、独特的优越性和应用前景。endprint
References
1 Rikken G L J A, Raupach E. Nature, 2000, 405: 932-935
2 Wang H D, Chu L Y. J. Membr. Sci., 2007, 297: 262-270
3 Maier N M, Franco P, Lindner W. J. Chromatogr. A, 2001, 906: 3-33
4 Lorin M, Delepee R, Maurizot J C, Ribet J P, Morin P. Chirality, 2007, 19: 106-113
5 Granados A G, Martinez A, Quiros R. Tetrahedron., 1999, 55: 8567-8578
6 Puglisi A, Benaglia M, Annunziata R, Bologna A. Tetrahedron. Lett., 2003, 44: 2947-2951
7 Miller L, Orihuela C, Fronek R, Murphy J. J. Chromatogr A., 1999, 865: 211-226
8 Zheng X Y, Yao T M, Zhu Y, Shi S. Biosens. Bioelectronics, 2015, 66: 103-108
9 Schiopu I, Iftemi S, Luchian T. Langmuir, 2015, 31: 387-396
10 Pietraszkiewicz M, Kozbial M, Pietraszkiewicz O. J Membr Sci., 1998, 138: 109-113
11 Yoshikawa M, Ooi T, Izumi J. J. Appl. Polym. Sci., 1999, 72: 493-499
12 Yang M, Zhao M, Xie S M, Yuan L M. J. Appl. Polym. Sci., 2009, 112: 2516-2521
13 Campion A, Kambhampati P. Chem. Soc. Rev., 1998, 27: 241-250
14 Lin W C, Jen H C, Chen C L, Hwang D F, Chang R, Hwang J S, Chiang H P. Plasmonics., 2009, 4: 178-192
15 XIE Yun-Fei, WANG Xu, RUAN Wei-Dong, SONG Wei, ZHAO Bing. Spectroscopy and Spectral Analysis, 2011, 31(9): 2319-2323
谢云飞, 王 旭, 阮伟东, 宋 薇, 赵 冰. 光谱学与光谱分析, 2011, 31(9): 2319-2323
16 Li P W, Zhang J, Zhang L, Mo Y J. J. Vib. Spectrosc., 2009, 49: 2-6
17 Lorenzo L R, Puebla R A A, Santos I P, Mazzucco S, Stéphan O, Kociak M, Marzn L M, Abajo F J Gl. J. Am. Chem. Soc., 2009, 131: 4616-4618
18 Dadosh T, Sperling J, Bryant G W, Breslow R, Shegai T, Dyshel M, Haran G, Joseph I B. ACS Nano, 2009, 3: 1988-1994
19 Lim D K, Jeon K S, Kim H M, Nam J M, Suh Y D. Nat. Mater., 2010, 9: 60-67
20 Lee S B, Mitchell D T, Trofin L, Nevanen T K , Sderlund H. Martin C R. Science, 2002, 296: 2198-2200
21 Liu Y, Li P, Xie L, Fan D Y, Huang S S. J. Membr. Sci., 2014, 453: 12-17
22 Yang Y, Krishna K, Joe G. Micropor. Mesopor. Mater., 2012, 153: 131-136
23 Jiao J Q, Tang J G, Wang G M, Wang Y, Huang L J, Huang Z, Liu J X, Zhu Y K, Belfiore L A. RSC Adv., 2015, 5: 60920-60925
24 SUN Lei, LIU Ai-Xin, HUANG Hong-Ying, TAO Xiao-Jun, ZHAO Yan-Bao, ZHANG Zhi-Jun. Acta Phys. Chim. Sin., 2011, 27: 722-728
孙 磊, 刘爱心, 黄红莹, 陶小军, 赵彦保, 张治军. 物理化学学报, 2011, 27: 722-728
25 LIU Yue, DAI Guo-Shuai, XIE Li, HUANG Shan-Sheng. Chinese J. Anal.Chem., 2014, 42(5): 623-628
柳 悦, 代国帅, 谢 利, 黄杉生, 分析化学, 2014, 42(5): 623-628endprint
