
埃洛石基阿司匹林表面印迹分离介质的可控制备及表征
2015-11-16 18:32苏立强周磊韩爽张维冰
分析化学 2015年11期
苏立强+周磊+韩爽+张维冰

摘 要 以环境友好的天然硅基纳米材料埃洛石为基质,阿司匹林为模板分子,丙烯酰胺为功能单体,采用可逆加成断裂链转移法(RAFT)制备阿司匹林表面印迹分离介质。紫外光谱结合Lamber-Beer理论模型从分子水平上证实印迹体系中模板分子与单体主要以1∶2形式结合成稳定复合物。通过红外光谱、透射电镜及静态吸附、选择性吸附对该印迹材料结构和吸附行为加以研究,结果表明:在埃洛石表面引入了一层厚度约为38 nm、均匀性良好的印迹层。与常规表面印迹及以硅胶为载体的印迹物相比,此印迹材料具有吸附容量高、印迹效果好的特点,印迹因子达到3.5。将印迹材料进一步用于模拟人体肠液扩散实验,印迹材料释药时间(12 h)是非印迹材料释药时间(6 h)的2倍,说明其具有较好释药效果,为可能的载药应用提供了基础数据。
关键词 分子印迹聚合物; 可逆加成断裂链转移; 埃洛石纳米管; 吸附特性
1 引 言
分子印迹聚合物(Molecularly imprinted polymer, MIP)[1]作为一种对目标分子具有显著识别的多功能型智能材料,已被广泛用于固相萃取、色谱分离、药物缓释等方面[2]。表面印迹[3]是将含有结合位点的MIP接枝到载体表面,与其它制备MIP方法相比,具有后处理简单、识别位点易接近、热力学平衡快等优点。但表面印迹多数属于自由基聚合,传统自由基聚合存在慢引发、快增长、易发生链终止和链转移等反应,决定了其过程难以控制。
可逆加成断裂链转移法(RAFT)[4]是使用广泛的活性-可控自由基技术,此法利用聚合物增长链与二硫酯化合物的可逆加成、加成物的可逆断裂以及链转移反应,可便捷地控制聚合物的分子量和分子量分布,在表面印迹自由基聚合中可实现活性聚合。RAFT与表面印迹技术结合的相关报道,文献[5,6]在球形硅胶、磁性硅球、氧化石墨烯表面制备印迹材料,取得了很好的结果;埃洛石纳米管(HNTs)与常用的载体碳纳米管、硅胶相比,表面既含有硅羟基,也含有铝羟基,且具有较大孔径,便于将识别材料引入到载体表面;另外HNTs价格便宜、无毒且生物相容性好。Pan等[7,8]以HNTs为基质,分别以2,4,6-三氯苯酚、2,4,5-三氯苯酚为模板分子,采用表面印迹法制备了两种MIP材料,均表现出较好的识别选择性。本实验以常用药物阿司匹林为模板分子,丙烯酰胺为功能单体,在HNTs表面采用RAFT法制备印迹材料,并对其结构和性能加以表征;并模拟人体肠液扩散实验,研究其作为药物载体的释药效果。2 实验部分
2.1 仪器与试剂
TU-1901型紫外可见分光光度计(北京普析通用仪器有限公司);AS380傅立叶变换红外光谱仪(美国尼高力公司);埃洛石(HNTs,河南郑州金阳光瓷器有限公司)。苯基溴化镁(PMB)、阿司匹林(Asp)、4-氯甲基苯基三氯硅烷(Sigma公司);二甲基乙二醇丙烯酸酯(EDMA,东京化成工业株式会社);偶氮二异丁腈(AIBN,北京化工厂);丙烯酰胺(AM)、甲基丙烯酸(MAA)、2-乙烯基吡啶(2-VP)均购自国药集团化学试剂有限公司,以上试剂均为分析纯。
2.2 苄基氯化HNTs的制备
称取1 g经HCl处理的HNTs,分散于20 mL甲苯溶剂中,加入2.5 mL 4-氯甲基苯基三氯硅烷,超声处理15 min后逐滴加入甲苯(2 mL)与三乙胺(1 mL)二者混合液,在惰性气体保护下,80℃回流搅拌24 h,产物经甲醇洗涤5次,60℃干燥,得到苄基氯改性的HNTs(HNTs-Cl)。
2.3 HNTs-RAFT的制备
取苯基溴化镁30 mL,分散于20 mL超干四氢呋喃中,预热至40℃,逐滴加入2 mL CS2,45℃反应2 h,再向混合液中加入苄基氯化HNTs 700 mg,在惰性气体保护下,60℃反应60 h,用1 mol/L HCl终止反应,产物依次用去离子水、乙醚各洗涤5次,60℃干燥,得到二硫代苯甲酸苄基酯链转移化HNTs(HNTs-RAFT)。
2.4 自组装印迹体系条件的筛选
2.4.1 结合力分析 以2-VP、MAA、AM为功能单体,使模板分子分别与上述功能单体按照物质量比为1∶4进行预聚合12 h。分别测定功能单体、模板分子及两者混合溶液的紫外吸收光谱,通过比较上述混合前后紫外吸收曲线的变化,选择功能单体。
2.4.2 印迹比例选择 以乙腈为溶剂,固定阿司匹林溶液的浓度,渐增功能单体用量,使二者物质量比分别为1∶0, 1∶2, 1∶4, 1∶6, 1∶8和1∶10,充分作用12 h后,测定上述溶液的紫外吸收光谱。
2.5 HNTs-MIP的制备
将模板分子Asp 0.0901 g(0.5 mmol)和功能单体AM 0.1422 g(2 mmol)溶于40 mL乙腈溶剂中,充分作用12 h,再加入HNTs-RAFT 700 mg、交联剂EDMA 1.9822 g(10 mmol)和引发剂AIBN 50 mg,通氮除氧15 min,60℃水浴反应24 h,产物用乙酸-甲醇 (20∶80,V/V)混合液索氏提取24 h,再用乙腈除去残留的醋酸,60℃干燥,得到印迹聚合物(HNTs-MIP)。非印迹聚合物(HNTs-NIP)制备方法同上,只是在反应体系中不加入Asp。
2.6 聚合物的释药行为研究
分别称取适量已吸附一定浓度阿司匹林的HNTs-MIP和HNTs-NIP,加入pH 6.8的人工肠液,利用紫外光谱法测其在不同时间的释药率。3 结果与讨论
3.1 印迹自组装体系条件的筛选
3.1.1 结合力分析 MIP的选择识别性是由模板分子与功能单体间结合力大小决定的,因此,优选合适的功能单体十分重要。采用紫外光谱法[4]系统研究Asp与功能单体2-VP, MAA和AM在乙腈中结合情况,结果表明,3种单体与模板均有作用,与其它两种单体相比,AM与模板分子结合力最大。这是由于碱性单体AM易与呈酸性的模板分子阿司匹林以分子间氢键形式结合成稳定复合物。虽然2-VP也呈碱性,但其结构较大,与阿司匹林结合时,存在空间位阻,结合受影响。因此,选择AM为单体。endprint
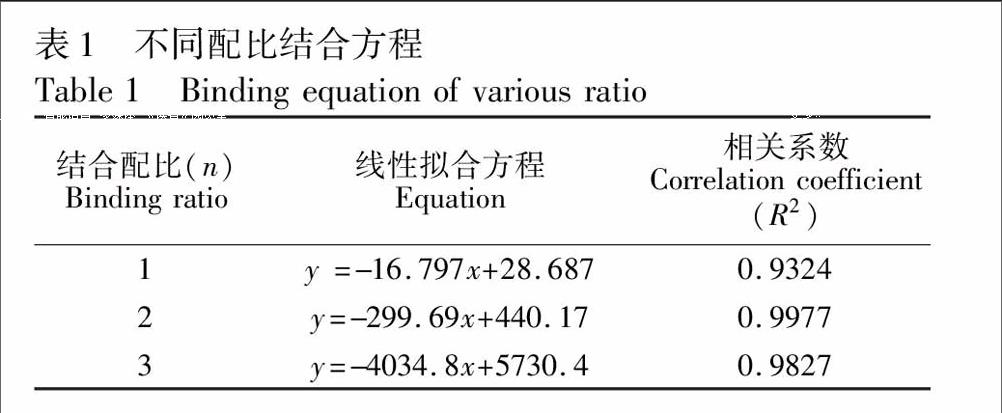
3.1.2 印迹比例分析 利用紫外光谱结合Lamber-Beer理论模型考察单体与模板比例对结合力的影响[9],结果如表1所示。n=1, 2, 3时,拟合方程均呈良好线性关系,其原因可能是在印迹过程中,Asp和AM有多个作用位点,二者可以1∶1、1∶2及1∶3形式结合成复合物,而当n=2时,线性相关系数最高,说明了Asp与AM以1∶2形式复合物最稳定;结合Asp、AM结构可以推测是丙烯酰胺分子中NH2、CO与乙酰水杨酸分子中COOH以分子间氢键形式结合成六元环状复合物,而另一个丙烯酰胺中NH2又与乙酰水杨酸分子中COOR的CO形成氢键。
理论上,Asp与AM主要以1∶2形式结合成稳定复合物,但在实验中,由于AM极易发生自聚,这就减少实际参与自组装印迹体系中AM用量,为确保Asp与AM充分结合,增加单体用量更为有利;根据文献[10]选择印迹比例1∶4。然而并不是二者比例越高越好,AM用量过高时,既能导致余下AM发生自身缔合,也会使未参与自组装印迹体系中功能残基增多,从而增加了低亲和结合位点数目,影响了MIP对Asp选择性识别能力。
3.2 HNTs-MIP结构表征
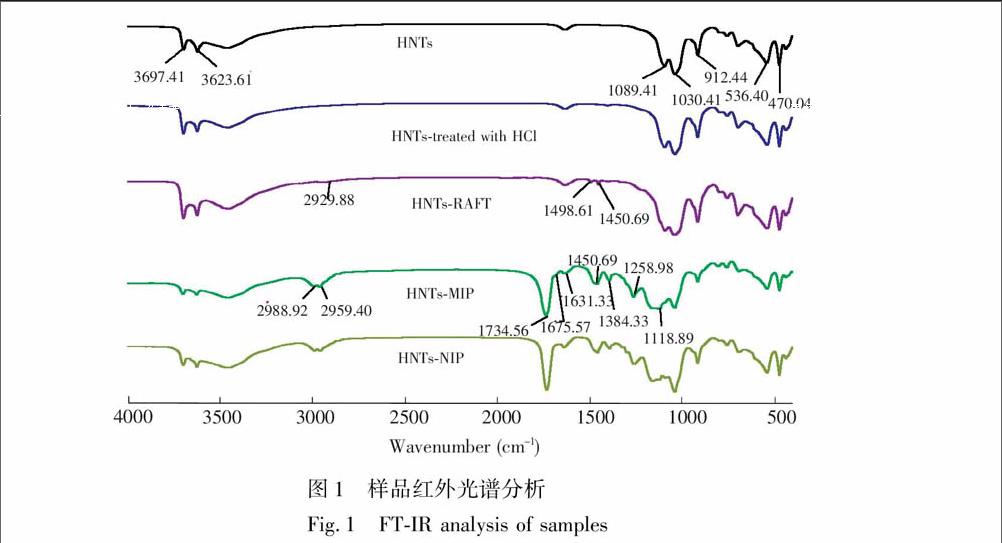
利用红外光谱研究了HNTs、HCl处理的HNTs, HNTs-RAFT, HNTs-MIP, HNTs-NIP的结构特征(图1),在HNTs中,3623.61和3697.41 cm
分别是铝羟基及硅羟基伸缩振动峰;而在HCl处理的HNTs中,上述两个硅铝羟基伸缩振动峰增强,说明经HCl酸化的埃洛石表面富含硅铝羟基,为埃洛石进一步改性接枝提供了条件。
是二硫代苯甲酸苄基酯中亚甲基和苯环特征峰,说明了RAFT试剂成功接枝到埃洛石表面;而在HNTs-MIP中,1384.33, 2959.40和2988.92 cm是交联剂中甲基的饱和碳氢特征峰,1118.89, 1258.98和1734.56 cm是交联剂中酯的碳氧键及羰基特征峰,1450.69 和1675.57cm是功能单体中酰胺的氮氢键及羰基特征峰,说明了印迹层成功包覆在埃洛石表面;另外, HNTs-NIP与HNTs-MIP主要特征峰十分吻合,说明Asp成功从印迹层中萃取下来,且没有破坏印迹层结构。此结果进一步证明了在印迹过程中,Asp与AM是通过非共价键进行自组装。结合二者分子结构可推断,其主要为氢键作用,与结合力分析结果吻合。
图2中HNTs(a)、HNTs-MIP(b)分别为埃洛石和埃洛石表面印迹物的透射电镜图。从图2可见,天然的埃洛石呈开口、双层、多壁纳米管状结构,且其内部有空腔,经分析,其壁厚约为29 nm。
埃洛石表面印迹聚合物后,开口、双层管状的形态未变,只是外层加厚,埃洛石表面包裹一层均匀的
图2 HNTs(a)和HNTs-MIP(b)的透射电镜图
Fig.2 TEM images of halloysite nanotubes (HNTs) (a) and HNTs-MIP(b)印迹层,反应后形成的聚合物形貌规整,这是由于RAFT聚合法的可控性,正是其优于传统自由基聚合法之处。规则、均匀的印迹层对于其选择、吸附等性能均是有利的。另外,图2b中HNTs-MIP壁厚约为67 nm,通过与HNTs壁厚进行对比,MIP平均厚度约为38 nm。
3.3 聚合物的吸附性能研究
图3 不同载体及方法的吸附量
Fig.3 Adsorption capacity of various carriers and methods
各称取8组20 mg印迹聚合物和非印迹聚合物,加入Asp不同浓度溶液,振荡后静止吸附24 h,计算二者吸附量。采用静态吸附法考察不同载体、不同方法制备聚合物的吸附性能(图3)。HNTs-MIP和Silica-MIP分别为采用RAFT表面印迹法在埃洛石和硅胶表面制备的印迹物,T-MIP为采用常规表面印迹法制备的印迹物, 由图可知,3种 印迹物MIP饱和吸附量均高于相应NIP吸附量,化学组份相同的材料有如此大的吸附差别,原因是二者在空间构型上具有显著差异,印迹材料存在与Asp空间构型和功能基团相吻合的印迹孔穴,该孔穴对Asp具有高度响应性,可实现对Asp专一性识别;而非印迹材料不含有这样的印迹孔穴,对Asp的识别主要是物理吸附[11];计算HNTs-MIP, Silica-MIP, T-MIP印迹因子(β=QMIP/QNIP)分别为3.5, 2.4, 1.7,发现HNTs-MIP, Silica-MIP除了饱和吸附量要远高于T-MIP之外,二者印迹因子也高于T-MIP。这是由于RAFT法可有效控制传统方法在聚合过程带来的慢引发、快增长、链终止及链转移等不可控因素,使印迹层厚度更加均匀,形成的印迹孔穴数目更多,从而提高其结合能力;另外还注意到,HNTs-MIP专一识别能力好于Silica-MIP,这主要是与硅胶相比,埃洛石表面既含有硅羟基,也含有铝羟基功能基团,且具有较大表面积,便于MIP的引入,印迹效果更好。
选取水杨酸(SA)和苯甲酸(BA)作为阿司匹林结构类似物,考察印迹物的选择性。称取3组HNTs-MIP和HNTs-NIP各20 mg,加入相同浓度阿司匹林、苯甲酸及水杨酸溶液,振荡后静止吸附24 h,测量上清液中上述3种苯系物的浓度,计算吸附量。经分析,Asp, SA, BA印迹因子β依次是3.5, 2.2, 1.6,由以上数据
图4 聚合物的释药曲线
Fig.4 Drug sustained release curve of polymer
HNTs-MIP负载药物量:78.28 mg/g (load drug amount of HNTs-MIP: 78.28 mg/g); 溶液pH: 6.8 (solution pH: 6.8)endprint
可知,HNTs-MIP对阿司匹林表现高度的选择识别性。
3.4 聚合物的释药行为研究
印迹物因其高度亲和性,用于药物缓释系统能提高载药量,增加药物的稳定性,克服了传统机械地将药物包覆胶囊里在释药过程中存在突释等缺点,使给药系统具有更持久的缓释周期[12]。本研究以常用药物阿司匹林为模板分子,HNTs-MIP为药物载体进行药物缓释实验研究,结果如图4所示,随给药时间增加,HNTs-MIP和HNTs-NIP释药率也随之增长,HNTs-NIP释药率几乎呈直线增长,而HNTs-MIP释药率则呈非线性增长,这是由于HNTs-NIP给药过程仅受扩散作用控制,缓释效果较差,而HNTs-MIP给药过程是由扩散作用和印迹效应二者共同决定,使药物模板Asp更好地在HNTs-MIP上选择性释放,以便维持人体血药浓度平衡,同时还减少患者服药次数,达到阿司匹林缓释目的。与HNTs-NIP释药时间(6 h)相比,HNTs-MIP释药时间长达12 h, 为其2倍,释药效果较好。
上述实验结果表明,在HNTs表面RAFT法制备的HNTs-MIP均匀性较好,对模板阿司匹林表现高度选择性以及较强的释药能力。
References
1 ZHU Li-Li, CAO Yu-Hua, CAO Guang-Qun. Chinese J. Anal. Chem., 2013, 41(11): 1724-1728
朱丽丽, 曹玉华, 曹光群. 分析化学, 2013, 41(11): 1724-1728
2 WANG Ying, HUANG Chun-Fang, LIANG Ru-Ping, QIU Jian-Ding. Chinese J. Anal. Chem., 2013, 41(5): 787-794
王 莹, 黄春芳, 梁汝萍, 邱建丁. 分析化学, 2013, 41(5): 787-794
3 Bi X D, Liu Z. Anal. Chem., 2014, 86(1): 959-966
4 Su L Q, Guo X L, Han S. Anal. Methods, 2014, 6: 2512-2517
5 Li Y, Zhou W H, Yang H H, Wang X R. Talanta, 2009, 79: 141-145
6 Li Y, Li X, Dong C K, Qi J Y, Han X J. Carbon, 2010, 48(12): 3427-3433
7 Pan J M,Yao H, Xu L C, Ou H X, Huo P W, Li X X, Yan Y S. J. Phys. Chem. C, 2011, 115(13): 5440-5449
8 Pan J M, Wang B, Dai J D, Dai X H, Hang H, Ou H X, Yan Y S. J. Mater. Chem., 2012, 22(8): 3360-3369
9 Cao H, Xu F, Li D X, Zhang X G, Yu J S. Res. Chem. Intermed., 2012, 39 (6): 6423-6428
10 SU Li-Qiang, LUAN Tian, GUO Xiao-Li. Chemistry, 2012, 75(10): 908-913
苏立强, 栾 田, 郭晓丽. 化学通报, 2012, 75(10): 908-913
11 LIU Lu-Kuan, YANG Wen-Ming, XU Wan-Zhen, ZHOU Zhi-Ping, LIU Hong, YAN Yong-Sheng. Chinese J. Anal. Chem., 2014, 42(2): 249-257
刘路宽, 杨文明, 徐婉珍, 周志平, 刘 鸿, 闫永胜.分析化学, 2014, 42(2): 249-257
12 LING Xia, LI Hong-Ping, GUO Juan, TANG You-Wen, LAI Jia-Ping. Acta Chim. Sinica, 2010, 68(1): 95-101
凌 霞, 李红萍, 郭 娟, 汤又文, 赖家平. 化学学报, 2010, 68(1): 95-101endprint
