
NOx和SO2在Cu2O(111)表面上同时脱除的DFT研究
2015-12-31 12:18范宝芸吕永康
山西化工 2015年6期
范宝芸, 王 凯, 吕永康*
(1.太原理工大学,山西 太原 030024;2.太原理工大学煤科学与技术教育部和山西省重点实验室,山西 太原 030024)
引 言
SO2和NOx的大量排放是空气质量持续恶化、酸雨危害日益严重的主要原因之一。煤炭的燃烧和汽车尾气是造成他们大量排放的主要根源。近年来,随着国家出台越来越严格的环境政策,人们开始重视对SO2和NOx排放的控制[1-2]。由于SO2和NOx的分别治理存在占地面积大、设备复杂、有二次污染等问题,难以满足日益严格的环保要求。因此,发展经济有效的脱硫脱硝一体化技术是国内外研究人员竞相开展的工作重点[3]。燃煤烟气脱硫脱硝一体化技术是将脱硫与脱硝技术组合为一套工艺流程,这样既简化工艺和设备,又结构紧凑、副产物少、建设和运行费用低[4]。
铜基催化剂因其较高的脱硫活性和脱硝活性以及较好的经济实用性得到了人们的广泛研究。其中,Cu2O是铜基催化剂中在实验条件下热稳定性最好的相,并且作为氧化物表面上催化反应的模型也得到了大量的研究[5-7]。Onsten等[8]通过研究发现,Cu2O能够较好地促进SO2的分解。使用DFT的方法,Sun 等[5,9]发现,Cu2O 体现出了对脱除NOx较好的催化活性。但是,查阅大量文献后我们发现,有关SO2和NOx在Cu2O上面共同脱除的报道较少。因此,很有必要研究SO2和NOx在Cu2O上面共同脱除的机理。
Cu2O(111)表面已经被证明是Cu2O的主要面[10]。本文采用Cu2O(111)表面作为底物,系统地研究了SO2和NOx在Cu2O(111)表面上的吸附以及他们之间的相互作用。采用DFT+U并结合周期性平板模型的方法研究了SO2和NOx在Cu2O(111)上的吸附构型和吸附能以及反应热和反应能垒等性质。弄清SO2和NOx在Cu2O(111)上相互作用的反应机理,将有助于科研工作者进一步改进SO2和NOx同时脱除工艺。
1 计算方法和模型
1.1 计算方法
为了系统地研究SO2和NOx在Cu2O(111)上面吸附和分解的能量和几何构型,我们使用VASP(vienna ab initio simulation package)软件包[11-12]和GGA-PAW[13-15]交换关联势进行 DFT+U 计算[16-17]。DFT+U是采用对Cu原子3d中的电子进行“+U”矫正的方法来弥补PBE计算过程的缺陷。计算时K点[18]全部采用3×3×1,平面波基组截断能均为400eV。优化时,力常数≤0.05hatree/Bohn时达到收敛要求。搜索过渡态时,采用NEB(nudged elastic band)方法[19-21],且所有的过渡态均通过虚频分析验证。当存在唯一虚频值时,即认为是过渡态。
1.2 表面模型
基于赤铜矿的晶胞构型[22][见图1a)],用超胞的方法做出了周期性的Cu2O(111)表面。对于Cu2O(111)表面而言,氧终止的无极性Cu2O(111)表面作为理想模型应用比较广泛,通常被用在理论[9,23-24]和实验[10,25]中来研究吸附性质、结构以及稳定性等。面上暴露着4个不同的表面原子:配位饱和的铜原子CuCSA,配位不饱和的铜原子CuCUS,配位饱和的氧原子OSUB,配位不饱和的氧原子OSUF,如图1b)所示。本文所用的模型是9层p(2×2)的周期性平板模型。在所有的计算中,最上面的那6层原子和要吸附的吸附质允许弛豫,下面3层原子是固定在体相上的。用15Å(1Å=10-10m下同)的真空层来分开周期性重复的平板模型。优化后,Cu2O的理论晶格常数为aCu2O=4.244Å,与实验值一致[26]。
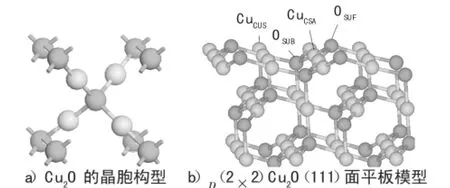
图1 Cu2O的晶胞构型和Cu2O(111)面平板模型
对于物质AB来说,AB的吸附能Eads定义如下:Eads=E(AB/slab)-E(slab)-E(AB)。其中,E(AB/slab)为吸附分子AB吸附在底物的总能量,E(slab)为清洁的底物能量,E(AB)为自由态分子AB的能量。对于反应AB=A+B来说,反应热ΔH计算通过如下公式:ΔH=E[(A+B)/slab]-E(AB/slab)。其中,E[(A+B)/slab]表示产物A+B共吸附在底物的总能量。活化能Ea计算公式为:Ea=E(TS/slab)-E(AB/slab)。其 中,E(TS/slab)表示过渡态分子吸附在底物上的能量。
2 结果与讨论
这一部分,我们首先提出了SO2和NOx在Cu2O(111)表面上相互作用的可能反应网络;然后,讨论它们在Cu2O(111)表面上的每一种可能反应中所涉及的吸附构型和吸附能;最后,搜寻每一个过程中的过渡态以及计算出每一个过程的反应热和能垒。
2.1 SO2和NOx在Cu2O(111)表面上相互作用的可能反应网络
SO2和NOx的相互作用开始于吸附在Cu2O(111)表面的SO2和NOx分子。吸附态的SO2和NOx分子进一步通过一系列的反应最后生成SO3或者S、NO2或者N2等易于捕集或者对大气无污染的物质。图2呈现了SO2和NOx在Cu2O(111)表面相互作用的可能反应网络图。

图2 Cu2O(111)表面上SO2和NOx可能的相互作用网络图
2.2 吸附分子及基团的结构与能量
对游离态的 SO2,S—O 的键长1.448Å,O—S—O键角119.1°,均与实验值一致[27]。优化后,SO2在Cu2O(111)面上最稳定的吸附构型如第3页图3a)所示,SO2垂直于Cu2O(111)表面。其中,SO2的S向上,SO2的1个氧与一配位铜相连;S—Ob键长从1.448Å延长至1.495Å,O—S—O键角从119.3°减少至115.8°,相应的吸附能为-85.867kJ/mol。
优化后,游离态SO中S—O的键长是1.201Å。SO在Cu2O(111)表面上最稳定的吸附构型如第3页图3b)所示。此时的SO平行于Cu2O(111)表面,且S与一配位铜相连。S—O的键长从1.201Å伸长至1.533Å,这个构型的吸附能大小是-170.287kJ/mol。
对S来说,在Cu2O(111)表面上最稳定的吸附构型是S吸附在“3Cu”位上,对应的构型图如第3页图3c)所示。此时的S吸附在3个铜原子上:1个CuCUS和2个CuCSA,这种计算结果和章日光等[6]的计算结果一致。CCUS—S和CuCSA—S的键长分别是2.098Å和2.331Å,对应的吸附能是-371.834kJ/mol。在这篇文章中,我们定义一种“3Cu”位:由1个CuCUS和2个CuCSA组成。这表明了周围原子对一配位铜原子能量贡献的重要性。
类似于S在Cu2O(111)表面上的吸附,经优化后,O在Cu2O(111)表面上也稳定地吸附在“3Cu”位上,如图3d)所示。CuCUS—O和CuCSA—O的键长分别是1.769Å和2.008Å。这样的计算结果也很好地与Chen等[28]的研究相吻合。吸附能大小为-453.938kJ/mol。
对于游离态的SO3,优化后,3个S—O的键长分别是 1.452、1.452 和 1.682 Å,与文献值一致[29-30]。SO3在Cu2O(111)表面上最稳定的吸附构型如图3e)所示。此时,SO3通过1个O吸附在一配位铜上面,另外2个O飘在2个相邻的二配位铜上。SO3中3个O组成的平面与整个表面平行,S在平面的上方。3个S—O的键长分别是1.536、1.506和1.503Å,3 个 O—S—O 的键角分别是109.1、108.9 和 110.9°,计 算 出 的 吸 附 能 为-92.331kJ/mol。
游离态的NO2,优化后,N—O的键长是1.215Å,O—N—O的键角是133.3°,都和实验值1.200Å和133°~134°一致[31]。NO2吸附在 Cu2O(111)面上最稳定的构型如图3f)所示,此时的NO2与Cu2O(111)表面垂直,NO2的N向上,其中的1个O与一配位铜CuCUS相连。以上结果与Sun等[5]的研究结果相似。N—O键长延长至1.246Å,O—N—O键角减少至123.7°,相应的吸附能为-152.342kJ/mol。
优化后,游离态的NO中N—O的键长是1.132Å,这与参考文献值1.120Å~1.170Å 一致[32]。NO在 Cu2O(111)表面上有氧端吸附和氮端吸附2种吸附构型。优化后,最稳定的吸附构型为NO以氮端吸附在一配位铜上面,如图3g)所示。此时,NO垂直吸附于Cu2O(111)表面,N—O的键长从1.132Å增长至1.179Å。这个构型吸附能的大小是-104.777kJ/mol。以上计算结果与Sun等[5]的研究结果一致。
优化后,游离态的N2O的构型是一条直线,O在这条直线的一端。O—N和N—N的键长分别是1.200和1.146Å,与文献值一致[33]。优化后,N2O在Cu2O(111)表面上最稳定的吸附构型如图3h)所示。从图3h)中可以看出,N2O按照O端吸附的方式倾斜地吸附在Cu2O(111)表面上。其中,N—N、O—CuCUS和 O—N的键长分别是1.140、2.011和1.216Å。这表明Cu2O(111)表面对N2O的O—N有较好的活化作用,这种活化有利于N2O下一步的分解。CuCUS—O—N的键角是119.6°,对应的吸附能是-117.320kJ/mol。
对游离态的N2,优化后,N—N的键长是1.055Å,和实验值1.431Å一致[34]。N2在Cu2O(111)表面上最稳定的吸附构型如图3i)所示。此时,N2垂直地吸附在一配位铜CuCUS上。N—N和N—CuCUS的键长分别是1.122Å和1.811Å,这与Sun等[5]的研究结果一致。计算出的吸附能是-91.333kJ/mol。
优化后,游离态的NOSO2构型如图3j)所示。此时,N—O已经断裂,N和O都连接到S上,并且都在SO2中O的对立侧。N和O之间的距离是1.492Å,Oa—S和N—S的键长分别是1.653Å和1.640Å,O—S—N 的键角是53.9°。NOSO2在Cu2O(111)表面上最稳定的吸附构型如图3j)所示。NOSO2中1个O与CuCUS相连,1个O和N飘向真空层,Oa与CuCSA相连。2个O—S的键长分别是1.433Å和1.490Å,Oa—S和 N—Oa的键长分别是1.581Å和1.697Å。

图3 Cu2O(111)表面上涉及到的分子和基团的吸附构型
2.3 SO2和NOx在Cu2O(111)表面上相互作用的热力学及动力学分析
对于反应R1而言,这个过程是SO2和NO2直接进行氧化还原反应的过程。该过程的初态(IS1)、TS1、末态(FS1)的构型图以及相应的势能图均见图4。从图4中可以看出,从SO2和NO2吸附在2个相邻CuCUS上的共吸附构型开始,这个路径的主要过程是NO2中N—Oa发生断裂,当N和Oa之间的距离达到1.835Å时,得到这个过程的过渡态。在TS1中,N—Oa已经发生断裂,S和Oa之间的距离从IS1中的3.550Å减小到1.959Å,N—Ob的键长为1.204Å。经过TS1之后,Oa链接在S上面,形成SO3。此时,S—Oa的键长是1.522Å。从势能图中可以看出,整个过程放出的热量是108.347kJ/mol,需要克服的能垒是121.372kJ/mol。
对反应R2,图4详细地展示了相关的势能图以及IS2、TS2的构型图。从图4中可以看出,这个路径的主要反应过程是吸附CuCUS上的NO在吸附于相邻CuCUS上的SO2的作用下,飘向SO2。N—CuCUS的键长从1.776Å逐渐增长至TS2中的4.293Å。在TS2中,N—O、S—O和N—S的键长分别是1.167、2.571和2.372Å。经过 TS2后,SO2和NO形成NOSO2,形成的NOSO2吸附在CuCUS—CuCSA桥位上。从势能图中可以看出,这个过程的能垒大小是269.469kJ/mol,并且需要吸收的热量是31.452kJ/mol。

图4 Cu2O(111)表面上SO2和NOx相互作用的势能图及初态、过渡态和末态的构型
对于反应R3,整个过程的IS3、TS3、FS2构型图和势能图都呈现在第5页图5中。从图5可以看出,整个过程从SO2和NO吸附在2个相邻的CuCUS上的共吸附构型开始,S—Oa逐渐增长,当S和Oa之间的距离达到1.621Å时,得到这个过程的TS3。经过TS3后,Oa和N相连,形成了FS3中的NO2。在TS3中,Oa飘向N,它们之间的距离从IS3中的3.581Å减小至2.050Å。从势能图中可以看出,整个过程中吸收112.592kJ/mol的热量,需要克服的能垒是221.808kJ/mol。
对于反应R4,这个过程的IS4、TS4、FS3的构型图和相应的势能图详见第5页图5。从前面的讨论可以知道,SO和NO都会优先吸附在CuCUS上,所以,我们选用上述共吸附构型作为本反应的IS4。选用S吸附在 “3Cu”位上同时NO2吸附在相邻CuCUS位上共吸附的构型作为该过程的FS4。从IS4开始,整个过程可以描述为:SO中的S—O断裂,经过过渡态TS4,解离的O飘向吸附状态的NO,生成NO2吸附在CuCUS上。从TS4的构型中可以看出,此时,S与O的距离增长至2.044Å,O和N之间的距离是2.793Å。经过TS4后,生成的S和NO2都吸附在相应的吸附位上,S和O之间的距离是3.779Å。由势能图可知,整个过程中吸收2.219kJ/mol的热量,需要克服的能垒大小是237.148kJ/mol。
对于反应R5,所涉及的IS5、TS5、FS4的构型图和势能图如第5页图5所示。从图5中看出,R5的反应过程是从吸附在同一个CuCUS上的2个NO开始,经过TS5,2个NO相互靠近,一个NO中的N—O断裂,另一个NO向N靠拢生成N2O,生成的O吸附在“3Cu”位上形成FS5。从能垒图中可以看出,整个过程需要吸收热量10.902kJ/mol,需要克服的能垒是101.593kJ/mol。在TS5中,断裂的N—O键长从1.190Å伸长至1.767Å,2个N之间的距离从2.721Å减小至1.803Å。在FS5中,断裂的N—O键长是3.620Å。
对反应R6,所涉及的TS6、FS5的构型图和势能图都呈现在第5页图5中。如图5所示,吸附在CuCUS上N2O中的N—O逐渐增长,当N—O的键长从1.216Å伸长至1.532Å时,得到了这个过程的TS6。在TS6中,N2漂浮在真空层中,解离的O与一个CuCUS相连,CuCUS—O的键长是1.832Å。FS5中生成的N2吸附在CuCUS上,O吸附在“3Cu”位上并且二者共用一个CuCUS。此时,N和O之间的距离是2.668Å。从能垒图中可以看出,整个过程需要吸收56.827kJ/mol的热量,需要克服的能垒是139.992kJ/mol。
对于反应R7,该过程中IS7、TS7的构型和势能图都呈现在图5中。如图5所示,从IS7开始,这个路径的过程可以描述为:经过TS7,吸附在“3Cu”上的O逐渐靠近SO2,最后O吸附在SO2的S上面,形成的SO3吸附在CuCUS上面。在TS7中,单独的O与原来 “3Cu”位中的CuCUS和一个CuCSA断开,O和S之间的距离从IS7中的4.574Å减小为2.622Å。原先SO2中的2个S—O的键长分别变成1.509Å和1.451Å。从该过程的势能图中可以看出,整个过程放出的热量是108.347kJ/mol,需要克服的能垒是121.372kJ/mol。
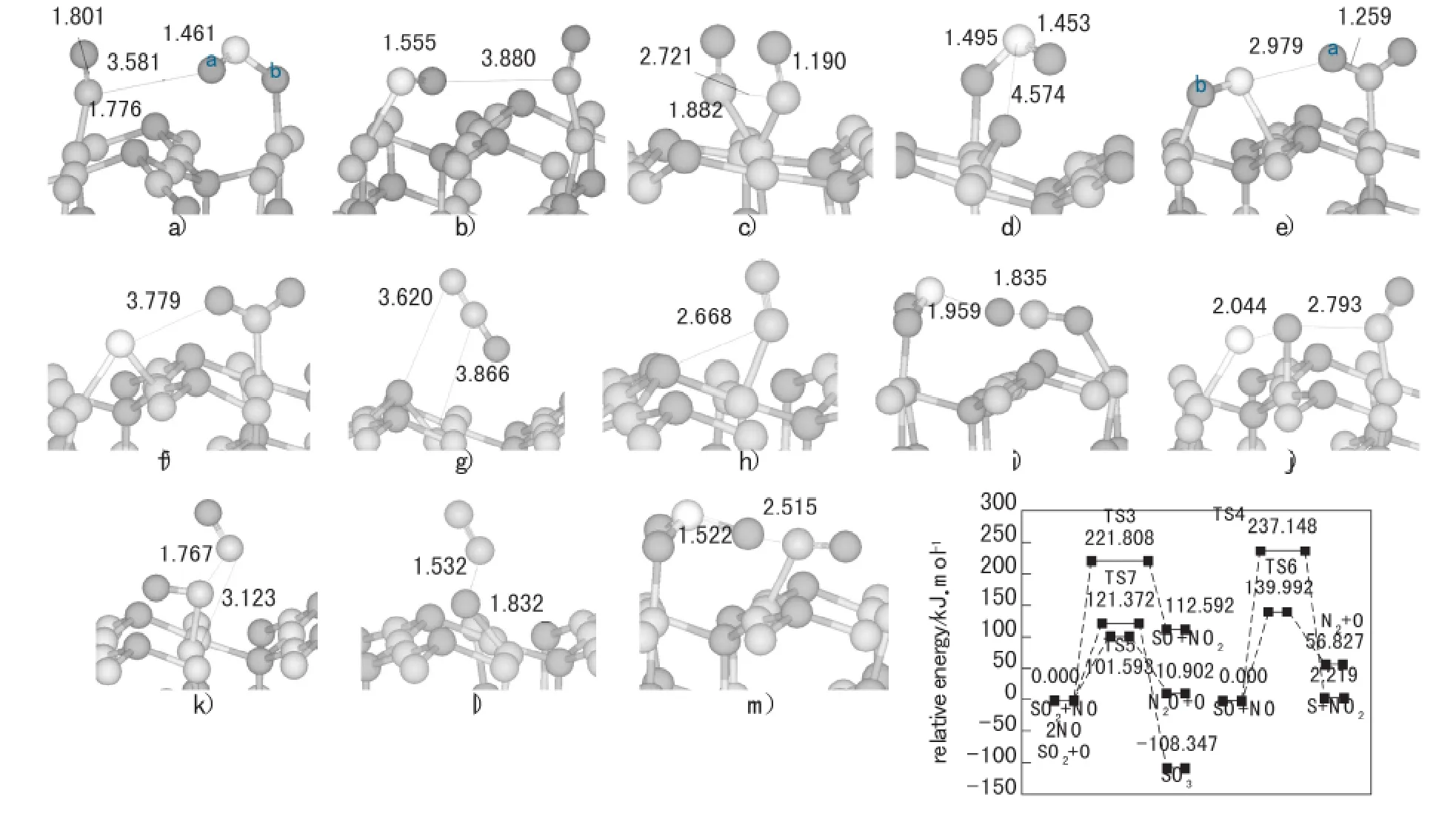
图5 Cu2O(111)表面上NOx与SO2相互作用过程中的势能图以及ISs、TSs、FSs的构型
3 结论
本文应用DFT结合周期性平板模型的方法,系统地研究了Cu2O(111)面上SO2和NOx共同脱除的反应机理。讨论了相关的活化能、反应热以及所有基元反应中过渡态的构型。计算结果表明,在Cu2O(111)表面上,NO2与SO2直接反应进而生成SO3和NO的过程需要克服的能垒是121.372kJ/mol。SO2与NO直接反应生成NOSO2时整个过程的能垒是269.469kJ/mol。但是,NO与SO2反应时,NO先以二聚体降解的方式得到O和N2O,然后N2O分解得到N2和O,最后用得到的O将SO2氧化成SO3。3个过程中的能垒分别是101.593、139.992和121.372kJ/mol。根据以上的研究结果得出了SO2和NOx在Cu2O(111)表面上共同脱除的机理:NO2首先与SO2反应生成SO3和NO,生成的NO与原先存在的NO一样,以二聚体的形式生成N2和O;然后,O将SO2氧化成SO3。该研究结果展示了NOx和SO2在Cu2O(111)表面上共同脱除的可行性,将为研究同时脱硫脱销提供理论上的支撑。
[1] Antony Stanislaus,Abdulazeem Marafi,Mohan S Rana.Recent advances in the science and technology of ultra low sulfur diesel(ULSD)production[J].Catalysis Today,2010,153(1):1-68.
[2] JoséA,Rodriguez,Jan Hrbek.Interaction of sulfur with well-defined metal and oxide surfaces:unraveling the mysteries behind catalyst poisoning and desulfurization[J].Accounts of Chemical Research,1999,32(9):719-728.
[3] 马双忱,马宵颖,郭天祥,等.微波改性活性炭用于烟气脱硫脱硝的实验研究[J].Journal of Fuel Chemistry and Technology,2010,38(6):55-57.
[4] 张翔宇.活性炭烟气脱硫脱硝集成工艺研究及废液燃烧烟气脱硫方案[D].天津:天津大学,2009:10-11.
[5] Sun Baozhen,Xu Xianglan,Chen Wenkai,et al.Theoretical insights into the reaction mechanisms of NO oxidation catalyzed by Cu2O(111)[J].Applied Surface Science,2014,316:416-423.
[6] Zhang Riguang,Liu Hongyan,Li Jingrui,et al.A mechanistic study of H2S adsorption and dissociation on Cu2O(111)surfaces:thermochemistry,reaction barrier[J].Applied Surface Science,2012,258(24):9932-9943.
[7] Anneli nsten,Mats Göthelid,Ulf O Karlsson.Atomic structure of Cu2O(111)[J].Surf Sci,2009,603(2):257-264.
[8] Annelio Nsten,Jonas Weissenrieder,Dunja Stoltz,et al.Role of defects in surface chemistry on Cu2O(111)[J].The Journal of Physical Chemistry:C,2013,117(38):19357-19364.
[9] Sun Baozhen,Chen Wenkai,Wang Xia,et al.A density functional theory study on the adsorption and dissociation of N2O on Cu2O(111)surface[J].Applied Surface Science,2007,253(18):7501-7505.
[10]Kirk H,Schulz,David F Cox.Photoemission and lowenergy-electron-diffraction study of clean and oxygendosed Cu2O(111)and(100)surfaces[J].Physical Review:B,1991,43(2):1610.
[11]Peter E Blöchl.Projector augmented-wave method[J].Physical Review:B,1994,50(24):17953.
[12]Georg Kresse,Jürgen Furthmüller.Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set[J].Physical Review:B,1996,54(16):11169.
[13]John P Perdew,Ja Chevary,Sh Vosko,et al.Atoms,molecules,solids,and surfaces:applications of the generalized gradient approximation for exchange and correlation[J].Physical Review:B,1992,46(11):6671.
[14]John P,Perdew,Wang Yue.Accurate and simple analytic representation of the electron-gas correlation energy[J].Physical Review:B,1992,45(23):13244.
[15]Ja White,Dm Bird.Implementation of gradient-corrected exchange-correlation potentials in car-parrinello total-energy calculations[J].Physical Review:B,1994,50(7):4954.
[16]Pang Xianyong,Wang Juanjuan,Wang Guichang.Selective oxidation of propylamine on oxygen-covered Au(111):a DFT study[J].Journal of Molecular Modeling,2012,18(8):3793-3804.
[17]王丙寅,于小虎,霍春芳,等.C2H4在Fe3C(100)表面吸附及脱氢裂解的密度泛函理论研究[J].催化学报,2013,35(1):28-37.
[18]Hendrik J,Monkhorst,James D Pack.Special points for Brillouin-zone integrations[J].Physical Review:B,1976,13(12):5188.
[19]Gregory Mills,Hannes Jónsson,Gregory K Schenter.Reversible work transition state theory:application to dissociative adsorption of hydrogen[J].Surf Sci,1995,324(2):305-337.
[20]Graeme Henkelman,Hannes Jónsson.Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points[J].The Journal of Chemical Physics,2000,113(22):9978-9985.
[21]Graeme Henkelman,Blas P,Uberuaga,et al.A climbing image nudged elastic band method for finding saddle points and minimum energy paths[J].The Journal of Chemical Physics,2000,113(22):9901-9904.
[22]Martin A Nygren,Lars Gm Pettersson,Alexander Freitag,et al.Theoretical models of the polar Cu2O(100)Cu+-terminated surface[J].The Journal of Physical Chemistry,1996,100(1):294-298.
[23]Mazharul M Islam,Boubakar Diawara,Vincent Maurice,et al.Bulk and surface properties of Cu2O:A firstprinciples investigation[J].Journal of Molecular Structure:Theochem,2009,903(1):41-48.
[24]Sun Baozhen,Chen Wenkai,Zheng Jinde,et al.Roles of oxygen vacancy in the adsorption properties of CO and NO on Cu2O(111)surface:Results of a first principles study[J].Applied Surface Science,2008,255(5):3141-3148.
[25]Lin J,Ja May,Sv Didziulis,et al.Variable-energy photoelectron spectroscopic studies of H2S chemisorption on Cu2O and ZnO single-crystal surfaces:HS-bonding to Copper(I)and Zinc(II)sites related to catalytic poisoning[J].Journal of the American Chemical Society,1992,114(12):4718-4727.
[26]Restori R,Schwarzenbach D.Charge density in cuprite,Cu2O[J].Acta Crystallographica Section:B:Structural Science,1986,42(3):201-208.
[27]William F Schneider,John Li,Kenneth C Hass.Combined computational and experimental investigation of SOxadsorption on MgO[J].The Journal of Physical Chemistry:B,2001,105(29):6972-6979.
[28]Shen Yaoyao,Feng Huitian,Chen Shougang,et al.Density functional theory study on the mechanism of CO sensing on Cu2O(111)surface:influence of the pre-adsorbed oxygen atom[J].Applied Surface Science,2014,288:452-457.
[29]Stanislav K Ignatov,Petr G Sennikov,Alexey G Razuvaev,et al.Ab-initio and DFT study of the molecular mechanisms of SO3and SOCl2reactions with water in the gas phase[J].The Journal of Physical Chemistry:A,2004,108(16):3642-3649.
[30]Evert Jan Meijer,Michiel Sprik.A density functional study of the addition of water to SO3in the gas phase and in aqueous solution[J].The Journal of Physical Chemistry:A,1998,102(17):2893-2898.
[31]Zhang Jiaxu,Liu Jingyao,Li Zesheng,et al.Theoretical study on the reaction mechanism of the methyl radical with nitrogen oxides[J].Journal of Computational Chemistry,2005,26(8):807-817.
[32] Wei Zhigang,Huang Xuri,Zhang Shaowen,et al.A theoretical study on the potential energy surface of the 3C2+ NO reaction[J].The Journal of Physical Chemistry:A,2004,108(32):6771-6777.
[33]Chen Wenkai,Sun Baozhen,Wang Xia,et al.The role of surface oxigen vacancy in N2O decompositoon on Cu2O(111)surface:a DFT study[J].Journal of Theoretical and Computational Chemistry,2008,7(2):263-276.
[34]Rodolfo Izquierdo,Leonardo J,Rodríguez,et al.Direct catalytic decomposition of NO with Cu-ZSM-5:ADFTONIOM study[J].Journal of Molecular Catalysis:A:Chemical,2011,348(1/2):55-62.
猜你喜欢
北京航空航天大学学报(2022年5期)2022-06-06
锦州医科大学报(2021年6期)2021-09-10
中学课程辅导·教学研究(2021年8期)2021-07-14
化学工业与工程(2021年3期)2021-06-25
燃料化学学报(2021年5期)2021-06-02
中学生天地(A版)(2020年3期)2020-04-10
中学教学参考·文综版(2018年9期)2018-10-23
电脑知识与技术(2018年3期)2018-03-21
海南医学(2017年7期)2017-05-11
哈尔滨理工大学学报(2017年1期)2017-04-08
