
1-取代苄氧基-3,6,7-三羟基-氧杂蒽酮的合成及NCI-H460细胞抑制活性研究
2016-01-19 01:38吴玮峰陆赛花刘秀凤
西北药学杂志 2016年1期
关键词:合成
张 晔,吴玮峰,陆赛花,刘秀凤,徐 峰
(上海市奉贤区中心医院药剂科,上海 201499)
1-取代苄氧基-3,6,7-三羟基-氧杂蒽酮的合成及NCI-H460细胞抑制活性研究
张晔,吴玮峰,陆赛花,刘秀凤,徐峰
(上海市奉贤区中心医院药剂科,上海201499)
摘要:目的研究苄基上不同的取代基对1-取代苄氧基-3,6,7-三羟基-氧杂蒽酮类化合物细胞毒性的影响。方法设计合成氧杂蒽酮类化合物8个,所有化合物均经过核磁确证,并进行了初步的细胞毒活性筛选。结果和结论所合成8个化合物属首次报道,其中6个化合物对NCI-H460肿瘤细胞产生抑制作用,目标产物1b、1d、1e和1f作用较强。
关键词:氧杂蒽酮;细胞毒性;合成
氧杂蒽酮类化合物是一类具有多种生物活性的化合物。对这一结构类型化合物的生物活性研究最早可以追溯到1968年,Bhattacharya课题组报道天然糖苷芒果苷具有利尿和强心作用[1]。随着研究的深入,在1972年一系列作为β-肾上腺素阻断剂的氧杂蒽酮化合物被发现[2]。同年,氧杂蒽酮类化合物阿克罗宁被发现具有体外抗肿瘤活性[3]。现已发现多种抗肿瘤活性比较好的化合物,如DMXAA[4]、普梭草素[5]和AH680[6]等。
氧杂蒽酮类化合物的构效关系研究也一直是热点[7],本课题组多年来一直致力于氧杂蒽酮类化合物的合成及生物活性研究,通过对该类化合物的活性普筛发现,在1,3,6,7-四羟基-氧杂蒽酮的1位羟基上引入烷基或苄基,其呈现一定的细胞毒活性。因此,本文以1,3,6,7-四羟基-氧杂蒽酮为先导化合物,在其1位羟基上选择性地引入不同的取代苄基,共设计合成了8个化合物,并对所合成的目标化合物进行了初步的细胞毒活性筛选(NCI-H460细胞),合成方法路线见图1。

图1目标化合物的合成路线
Fig.1 Synthetic route of targeted compounds
1仪器与材料
核磁共振氢谱采用Bruker Spectmspin AC-P300型核磁共振仪进行测定,选用DMSO-d6为溶剂,TMS为内标。熔点测试采用毛细管法,并在Yamato model MP-21型熔点测定仪上进行,温度未经校正。红外光谱采用Bruker Vector 22型红外光谱仪进行测定。所用试剂均为市售分析纯。
2合成实验
核磁共振氢谱采用Bruker Spectmspin AC-P300型核磁共振仪进行测定,选用DMSO-d6为溶剂,TMS为内标。熔点测试采用毛细管法,并在Yamato model MP-21型熔点测定仪上进行,温度未经校正。红外光谱采用Bruker Vector 22型红外光谱仪进行测定。所用试剂均为市售分析纯。
参考文献2.11,3,6,7-三甲氧基-氧杂蒽酮的合成方法[8-9]制备。
2.1.1制备2,4,5-三甲氧基苯甲酰氯(3)在三颈瓶中放入2,4,5-三甲氧基苯甲酸250 g(1.2 mol),同时室温下搅拌缓慢滴加氯化亚砜共计300 mL,滴加完成后继续加热反应至回流后4 h。待反应完毕,继续加入甲苯150 mL,并蒸馏共沸物直至无液体蒸出,随后冷却反应物产生白绿色固体,减压干燥后的粗品。最后用甲醇重结晶得2,4,5-三甲氧基苯甲酰氯237 g,收率达到87%,熔点:133~138 ℃。(文献值:收率:90%,熔点:135~138 ℃)
2.1.2制备(2-羟基-4,5-二甲氧基苯基)-(2’,4’,6’-三甲氧基苯基)-甲酮(5)在含有300 mL乙醚的三颈瓶中放入化合物(3)25 g(0.11 mol)以及1,3,5-三甲氧基苯(4)20 g(0.12 mmol),搅拌后产生悬浮液体。并在0 ℃冰浴条件下搅拌,缓慢加入60 g三氯化铝进行反应。加入三氯化铝后持续搅拌,室温下反应48 h。将反应液在搅拌状态下缓慢倒入浓盐水和冰水的混合物中,除去乙醚,分3次用甲苯400 mL提取,随后合并甲苯提取液,用无水硫酸钠进行干燥,干燥液蒸馏回收,得到油状产物。加入少量甲醇搅拌析出黄色固体,过滤得粗品,乙醇重结晶得化合物(5)31.4 g,收率达到90%,熔点:167~169 ℃。(文献值:收率:88%,熔点:167~169 ℃)
2.1.3制备1,3,6,7-四甲氧基-氧杂蒽酮(6)将化合物(5)34.8 g(0.1 mol)放入三颈瓶中,在室温条件下加入纯水225 mL,吡啶300 mL和四丁基氢氧化铵50 mL,加热至回流后继续反应,持续反应10 h后滴加浓盐酸,逐步调节溶液pH至弱酸性,采用二氯甲烷溶液分3次萃取后合并萃取液,将萃取液用水调节pH至中性,萃取液加入无水硫酸钠干燥,干燥液蒸馏出二氯甲烷后可得黄色固体,用甲醇重结晶得到白色目标产物,即化合物(6)27.4 g,收率为86%,熔点:201.1~203.3 ℃。(文献值:收率:87%, 熔点:200~203 ℃)
1HNMR (300 MHz, DMSO-d6, TMS):δ7.85 (s,1H), 7.62 (s,1H),7.42 (d,1H,J=2.1 Hz),7.28 (s,1H,J=2.1 Hz),2.34 (s,3H),2.35(s,3H),2.38(s,3H),2.37(s,3H).13C-NMR(100 MHz,CDCl3):δ174.60,165.76,161.23,161.14,154.49,150.85,146.58,117.05,106.00,106.01,99.08,95.82,91.22,55.55,54.90 ESI,Calcd. for C17H17O6+,[M+H]+,317.10,Found,317.07。
2.1.4制备1,3,6,7-四羟基-氧杂蒽酮(7)在三颈瓶中放入31.6 g(0.1 mol)化合物(6)和100 mL苯酚,室温条件下加入氢碘酸100 mL,加热反应至160 ℃产生回流持续反应12 h,随后将反应液在搅拌状态下缓慢倒入500 mL硫代硫酸钠饱和溶液中,产生大量黄色固体。过滤反应液得到黄色粗品后经甲醇重结晶得到黄色固体,即化合物(7)22.1 g,收率:87%,熔点:>250 ℃。1HNMR(300 MHz,DMSO-d6,TMS):13.13(1H,s),10.80(1H,s),10.76(1H,s),9.74(1H,s),7.32(1H,s),6.81(1H,s),6.37(1H,s),6.19(1H,s)。
2.1.5制备1-羟基-3,6,7-三甲氧甲基-氧杂蒽酮(8)在茄形瓶中加入化合物(7)26 g(0.1 mol),并加入150 DMF搅拌至溶解,室温下缓慢逐滴加入氯甲醚10 mL和DIEPA 10 mL。室温下继续搅拌反应,20 h后停止,将反应液缓慢倒入500 mL氯化铵饱和溶液,搅拌后有溶液中析出大量固体。过滤并干燥后得到固体,将固体经柱层析(石油醚∶乙酸乙酯为10∶1)可得到浅黄色固体(8)29.3 g,收率为75%,熔点:148.2~150.1 ℃。1HNMR (300 MHz,DMSO-d6,TMS):13.15 (1H,s),5.42 (2H,s),5.31 (2H,s),5.38 (2H,s),3.46(9H,s)
2.2目标化合物的合成
2.2.1制备1-(取代苄氧基)-3,6,7-三甲氧甲醚基-氧杂蒽酮(9a-h)在茄形瓶中加入化合物(8)0.39 g(0.01 mol),加入DMF 20 mL搅拌至溶解,室温条件下加入不同的取代氯苄150 mL,无水碳酸钾150 mg,搅拌10 h后将反应液倒入100 mL水中,搅拌后将析出的固体过滤后用少量乙醚和丙酮进行洗涤,烘干可得白色粉末。
2.2.2制备1-(取代苄氧基)-3,6,7-三羟基-氧杂蒽酮(1a-h)在茄形瓶中加入9a-h(5 mmol)并缓慢加入50 mL甲醇和10 mL 4 mol·L-1的盐酸,100 ℃回流条件下反应4 h。反应完毕后将溶剂蒸干,产生的固体用少量丙酮洗涤,烘干得产物(1a-h)。
2.2.3其他目标化合物均按照1.2.2项下方法合成,8个目标化合物经光谱确证结构,其理化及光谱数据见表1。
3药理实验
将0.2 mL胰蛋白酶消化液加入到NCI-H460细胞中,消化液使贴壁细胞脱落,制成细胞悬液。细胞计数,稀释细胞密度至5×104个·mL-1。将细胞悬液接种于96孔板上,每孔180 μL,置于恒温CO2培养箱中培养24 h。换液后加入受试药物,每孔20 μL,培养48 h。将MTT加入96孔板中,每孔20 μL,培养箱中孵育4 h。吸去上清液,加入DMSO,每孔150 μL,并在平板摇床上振摇5 min。在波长为490 nm 处用酶联免疫检测仪测定每孔的吸光度值,计算细胞抑制率[10-11]。以紫杉醇作为阳性对照,并用回归法求出各目标化合物的GI50。体外抑制NCI-H460肿瘤细胞数据见表2。

表1目标化合物的结构、熔点、产率和氢谱数据
Tab.1 The yield、melting point and1H-NMR data of the target compounds

化合物取代基R熔点/℃产率/%1H-NMR(DMSO-d6)δ1a2-F>25076.510.96~10.75(3H,b),7.62(1H,d),7.59~7.47(2H,m),7.37(1H,s),7.16(1H,m),6.79(1H,s),6.43(2H,m),5.20(2H,s).1b3-F247.1~248.396.110.91~10.64(3H,b),7.63(1H,d),7.62~7.37(3H,m),7.17~7.13(1H,m),6.80(1H,s),6.43(2H,m),5.19(2H,s).1c3-Et234.2~235.686.410.68(3H,b),7.73(1H,s),7.46~7.45(2H,d),7.28~7.25(2H,m),6.93(1H,s),6.27~6.16(2H,m),5.09(2H,s),2.54~2.48(2H,q),1.19~1.12(3H,m).1d4-Cl227.6~229.184.510.87~10.84(3H,b),7.70(2H,d),7.50(2H,d),7.49(1H,s),6.80(1H,s),6.43(2H,m),5.17(2H,s).1e3-Cl221.3~222.883.610.85~10.82(3H,b),7.58~7.55(1H,d),7.46~7.44(2H,m),7.73(1H,s),7.37(1H,m),7.12(1H,s),6.40(2H,s),5.20(2H,s).1f4-Br245.0~246.154.310.89(1H,b),10.87(1H,b),9.71(1H,b),7.62(4H,s),7.36(1H,s),6.81(1H,s),6.46~6.40(2H,m).1g2,4-diCl239.2~240.882.310.69(3H,b),7.33(1H,s),6.77(1H,m),6.38~6.30(2H,m),6.09~6.00(1H,m),5.75(1H,d),5.68(1H,d),4.60(2H,s).1h4-Et230.1~231.762.510.74(3H,b),7.75(1H,s),7.46~7.42(2H,m),7.24~7.22(2H,m),6.89(1H,s),6.28~6.18(2H,m),5.08(2H,s).
表2化合物体外抑制NCI-H460肿瘤细胞数据
Tab.2 The inhibitory effect of the synthesized compounds on NCL-H460invivo
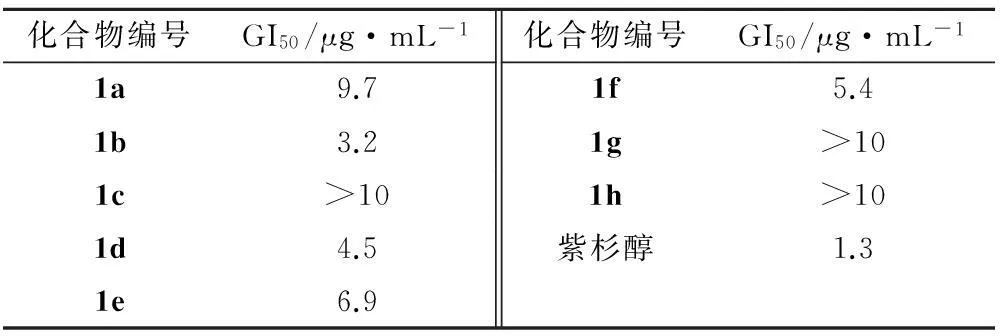
化合物编号GI50/μg·mL-1化合物编号GI50/μg·mL-11a9.71f5.41b3.21g>101c>101h>101d4.5紫杉醇1.31e6.9
4结论
经过初步的体外抗NCI-H460肿瘤细胞实验,所合成化合物中,多数化合物对NCI-H460肿瘤细胞有抑制作用,化合物1b、1d、1e和1f的GI50值分别为3.2,4.5,6.9和5.4 μg·L-1。根据药理活性数据得出,氧杂蒽酮-1位引入取代苄基可以使该类化合物产生抑制NCI-H460肿瘤细胞生长的作用,但鉴于目标化合物的数目有限,其构效关系还有待在以后的研究中进一步探讨。
[1]Finnegan R A,Stephani R A,Ganguli G,et al. Occurrence of mangiferin in Hiptage madablota geartn [J]. J Pharm Sci,1968,57(6):1039-1040.
[2]Da Re P, Primofiore G P,Bertelli A. Adrenergic blocking agents of the chromone and xanthone groups. II. Propanolol type derivatives [J]. J Med Chem,1972,15(8): 868-869.
[3]Schneider J,Evans E L,Grunberg E,et al. Synthesis and biological activity of acronycine analogs [J]. J Med Chem,1972,15(3):266-270.
[4]Jameson M B,Thompson P I,Baguley B C,et al. Clinical aspects of a phase I trial of 5,6-dimethylxanthenone-4-acetic acid (DMXAA),a novel antivascular agent [J]. Br J Cancer,2003,88(12):1844-1850.
[5]Terrance T,Schwaebe M K,Whitten J P,Process to prepare psorospermin. W.O. Patent 2004/019888,2004.
[6]Sylvia V L,Del Toro F Jr,Hardin R R,et al. Characterization of PGE(2) receptors (EP) and their role as mediators of 1α,25-(OH)(2)D(3) effects on growth zone chondrocytes [J]. J Steroid Biochem Mol Biol ,2001,78(3):261-274.
[7]程亚涛,徐昕,王鹏龙,等. 9种黄酮类化合物对肿瘤细胞的抑制活性及构效关系研究[J].西北药学杂志,2014,29(2):187-190.
[8]Lippert J W. 3rd. Vascular disrupting agents [J]. Bioorg Med Chem,2007,15(1): 605-615.
[9]Jean-Jacques Helesbeux,Olivier Duval, Caroline Dartiguelongue,et al. Synthesis of 2-hydroxy-3-methylbut-3-enyl substituted coumarins and xanthones as natural products. Application of the chenck ene reaction of singlet oxygen with ortho-prenylphenol precursors [J].Tetrahedron,2004,60(10):2293-2300.
[10]Monks A,Scudiero D,Skehan P,et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines [J]. J Natl Cancer Inst,1991,83(11): 757-766.
[11]田梅.Src激酶抑制剂对人肺癌顺铂耐药细胞A549/DDP多药耐药性的影响及机制[J].西北药学杂志,2013,28(2):170-174.
·化学·
Synthesis of 1-substituted benzyl-oxy-3,6,7-trihydroxyl xanthone with NCI-H460 cells inhibitory activity evaluation
ZHANG Ye,WU Weifeng,LU Saihua,LIU Xiufeng,XU Feng(Department of Pharmacy, Shanghai Fengxian Central Hospital,Shanghai 201499,China)
Abstract:Objective To study the influence on cytotoxicity of 1-substituted benzyl-oxy-3,6,7-trihydroxyl xanthone with different benzyl groups. MethodsEight target compounds were designed and synthesized. Their structures were confirmed by1H-NMR spectra. Resultsand ConclusionAll of the target compounds were reported for the first time. The results of preliminary pharmacological test showed that all the target compounds exhibited potent NCI-H460 cell inhibitory activity,of which the compound 1b,1d,1e and1f showed the highest activity.
Key words:xanthone;cytotoxicity;synthesis
收稿日期:(2015-07-09)
中图分类号:R914.5
文献标志码:A
文章编号:1004-2407(2016)01-0083-03
doi:10.3969/j.issn.1004-2407.2016.01.025
猜你喜欢
现代商贸工业(2016年14期)2016-12-27
安徽理工大学学报·自然科学版(2016年4期)2016-12-23
考试周刊(2016年85期)2016-11-11
农业与技术(2016年15期)2016-11-09
科技视界(2016年18期)2016-11-03
科教导刊·电子版(2016年19期)2016-08-19
科技视界(2016年15期)2016-06-30
科技视界(2016年10期)2016-04-26
科技视界(2016年9期)2016-04-26
科技视界(2016年3期)2016-02-26
