
常山酮中间体的合成工艺改进
2016-02-25 05:47王超杰宋卫强
合成化学 2016年1期
王超杰, 万 阳, 宋卫强, 陆 群
(西南交通大学 生命科学与工程学院,四川 成都 610031)
·制药技术·

通信联系人: 陆群,副教授, E-mail: 1607520335@qq.com
常山酮中间体的合成工艺改进
王超杰, 万阳, 宋卫强, 陆群*
(西南交通大学 生命科学与工程学院,四川 成都610031)
摘要:以2-乙酰呋喃为起始原料,经扩环、甲基化、去质子化、亲核加成、缩酮化、Rh/C催化氢化及水解等反应合成了常山酮中间体——1-(3-甲氧基哌啶-2-基)丙酮,总收率40.2%,其结构经1H NMR和ESI-MS确证。
关键词:2-乙酰呋喃; 常山酮; 中间体; 1-(3-甲氧基哌啶-2-基)丙酮; 合成; 工艺改进
常山酮(Chart 1)主要存在于植物常山和伞形秀球等植被中,临床研究结果表明,其具有较高的抗疟活性,同时具有抑制Ⅰ型胶原合成、辅助性T细胞17的分化和T细胞因子的产生等生物活性[1-5]。目前主要用于球虫病和隐孢子虫病的预防和治疗,由于其低残留、杀虫效果好及耐药性强等优点已成为治疗禽类球虫病的首选药物[6-8]。

Chart 1
常山酮的全合成少有报道,国内尚未实现工业生产,目前主要依托进口和从常山块根提取获得[9]。其合成的难点和关键在于哌啶环中间体——1-(3-甲氧基哌啶-2-基)丙酮(1)的合成。美国氰胺公司的Baker研究小组[10-11]在合成常山碱过程中首次合成了该中间体,并以此为基础合成常山酮,于上世纪60年代在美国作为抗球虫药上市。该合成路线的最大优点是原料易得,不足之处在于线路过长、收率低、甲基化后处理用到镉盐和贵金属催化剂Yb(OTf)3等。随后Donald小组和沈昌茂小组[12-13]先后对其进行了优化。
本文以文献[12-13]方法为基础,对1的合成工艺进行了优化。以2-乙酰呋喃(2)为起始原料,经高温高压扩环、甲基化、质子化、亲核加成、水解、缩酮化及Rh/C催化氢化等反应合成了1(Scheme 1),其结构经1H NMR和ESI-MS确证。

Scheme 1
1实验部分
1.1 仪器与试剂
SWG-2型自动恒温旋光仪(温度未校正);Varian INOVA-400型核磁共振仪(CDCl3为溶剂,TMS为内标);Waters Quattro Premiter XE型质谱仪;岛津LC-20AT型液相色谱仪。
所用试剂均为分析纯或化学纯。
1.2 合成
(1) 2-甲基-3-羟基吡啶(3)的合成
在500 mL高压反应釜内依次加入2 13.0 g(0.12 mol),三乙基苄胺树脂1.0 g和甲醇150 mL,于室温搅拌10 min使2完全溶解;再加入25%~28%氨水150 g(2.38 mol),密闭反应釜,于170 ℃(3.0~3.2 MPa)反应9 h。冷却至室温,加入活性炭30 g,于90 ℃回流反应30 min。趁热过滤,滤液减压浓缩除去甲醇和氨,剩余物用10%盐酸(约30 mL)调至pH 3,用乙酸乙酯(3×100 mL)萃取,水相用10%氢氧化钠溶液(50 mL)调至pH 8~9,用乙酸乙酯(3×100 mL)萃取,合并萃取液,浓缩得淡黄色粉末,用苯(2×50 mL)洗涤,干燥得类白色粉末3 10.3 g,收率72%, m.p.166~169 ℃(收率45%, m.p.168~170 ℃[14]);1H NMR(DMSO-d6)δ: 9.76(s, 1H, OH), 7.89(d,J=4.4 Hz, 1H, 6-H), 7.10(d,J=8.0 Hz, 1H, 4-H), 7.03(dd,J=4.8 Hz, 9.6 Hz, 1H, 5-H), 2.33(s, 3H, CH3); ESI-MSm/z: 110.06 {[M+H]+}。
(2) 2-甲基-3-甲氧基吡啶(4)的合成
在反应瓶中加入DMSO(依次用分子筛和氯化钙除水)500 mL,搅拌下油浴升温至130 ℃,加入甲醇钾40.6 g,搅拌使其溶解;加入3 50.0 g(0.46 mol),升温至170 ℃,反应20 min;分批加入氯化四甲铵65.0 g(0.59 mol),回流反应12 h(尾气用5%盐酸500 mL吸收)。冷却至室温,用20%硫酸65 mL调至pH 1,减压回收DMSO。剩余物(褐色黏稠状)用水(100 mL)溶解,用10%氢氧化钠溶液(200 mL)调至pH 10,用乙酸乙酯(3×200 mL)萃取,合并萃取液,减压浓缩后精馏得无色透明油状液体4 61.7 g,收率87%(75%[13]), b.p.40~42 ℃(27 Pa);1H NMRδ: 8.06~8.07(m, 1H, 6-H), 7.06~7.11(m, 2H, 4,5-H), 3.83(s, 3H, OCH3), 2.47(s, 3H, CH3); ESI-MSm/z: 124.07{[M+H]+。
(3) 1-(3-甲氧基吡啶-2-基)丙酮(5)的合成
将4 26.0 g(0.211 mol)溶于无水四氢呋喃(50 mL)中,冰盐浴冷却(-10~-5 ℃),搅拌下缓慢滴加正丁基锂16.0 g(0.25 mol)的正己烷(125 mL)溶液,滴毕,于室温反应2 h。冷却至0 ℃,缓慢滴加无水乙腈20 mL,控制温度<10 ℃反应过夜。加水20 mL,用10%盐酸酸化至pH≈1,减压回收THF,剩余物用10%NaOH溶液调至pH 10,用乙酸乙酯(3×200 mL)萃取,合并萃取液,浓缩后减压精馏得淡黄色油状液体5 24.0 g,收率69%(54%[13]), b.p.92~94 ℃(27 Pa);1H NMRδ: 8.14~8.15(m, 1H, 6-H), 7.15~7.20(m, 2H, 4,5-H), 3.95(s, 2H, CH2), 3.82(s, 3H, OCH3), 2.20(s, 3H, COCH3); ESI-MSm/z: 166.08{[M+H]+}。
(4) 1-(3-甲氧基吡啶-2-基)丙酮缩乙二醇(6)的合成
在反应瓶中依次加入5 15.0 g(0.09 mol),对甲苯磺酸18.6 g,乙二醇34.0 g(0.54 mol)和甲苯170 mL,搅拌下于120 ℃反应6 h。冷却至室温,加入10%氢氧化钠溶液200 mL,于室温搅拌10 min,分液,水相用乙酸乙酯(3×150 mL)萃取,合并有机相和萃取液,减压浓缩,浓缩液减压精馏得淡黄色液体6 18.2 g,收率96%(76.3%[13]), b.p.100~110 ℃(33 Pa);1H NMRδ: 8.18~8.19(m, 1H, 6-H), 7.13~7.14(m, 2H, 4,5-H), 3.93~3.97(m, 4H, CH2CH2), 3.86(s, 3H, OCH3), 3.23(s, 2H, CH2), 1.40(s, 3H, CH3); ESI-MSm/z: 232.03{[M+Na]+}。
(5) 1的合成
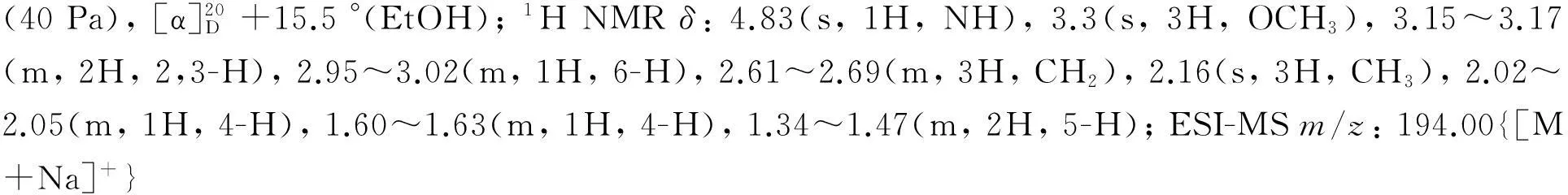
2结果与讨论
2.1 3的合成工艺优化
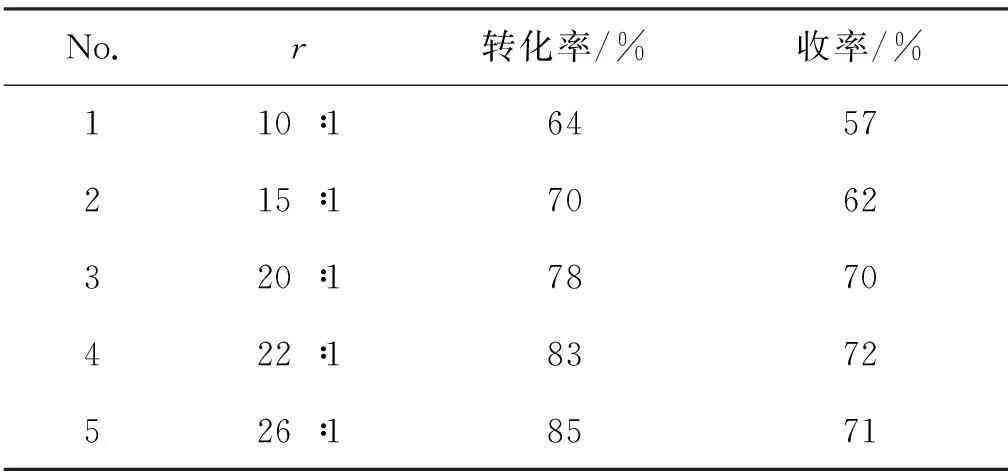
在3的合成中,结合文献[14-15]报道,2 0.12 mol,未加催化剂,其余反应条件同1.2(1),重点考察了原料配比[r=n(氨水) ∶n(2)]和反应温度对反应的影响,结果分别见表1和表2。由表1可见,随着r增大,3收率逐渐提高,r=22 ∶1时收率最大(72%)。故最佳r为22 ∶1。
表1r对3收率的影响*
Table 1 Influence ofron the yield of 3
No.r转化率/%收率/%110∶16457215∶17062320∶17870422∶18372526∶18571
**2 0.12 mol, 其余反应条件同1.2(1);r=n(氨水) ∶n(2)
由表2可见,转化率随温度升高而增大,但收率随温度变化呈现先增大后减小趋势,于170 ℃左右达到最佳(72%)。
在上述条件下,当加入文献报道的氯化铵和氯化锌等催化剂时并不能使收率明显提高,但加入新型催化剂三乙基苄胺树脂时,反应时间缩短至9 h。

表2 反应温度对3收率的影响
**r=22 ∶1,其余同表1
在3的合成中,后处理是该步反应的难点。文献报道多用重结晶的方法,但该方法周期长,收率低。本文根据原料、产物及副产物[3-羟基-6-氨基-3,5-己二烯-2-酮(7)和2-乙酰吡咯(8)](Chart 2)的理化性质差异,提出了利用酸化-碱化-洗涤的方法纯化3。在酸性条件下,由于吡咯环的氮原子上的电子均参与了环共轭,无多余的孤对电子,结合质子能力弱,使得7具有弱酸性,而呋喃环在弱酸条件下也是稳定的,而副产物8和产物3则会成盐,萃取分离出7和2,水相碱化后8和3重新游离出来,萃取浓缩,进而利用8和3极性差异,以苯洗涤除去副产物8,得产物3。该方法较重结晶法具有快速、省时和原料回收率高等优点。

Chart 2
2.2 4的合成工艺优化
文献[12-13,16]报道以具有挥发性、高毒性及致癌性的碘甲烷、硫酸二甲酯或仲氮甲烷等作为甲基化试剂,并且由于吡啶氮杂原子具有较强电负性,会与RO-离子形成竞争效应,上述几种甲基化试剂均会产生氮甲基化副产物,不利于产物的纯化。本文采用了鲜有报道的四甲基氯化铵作为甲基化试剂。该试剂是一种非挥发性结晶固体(m.p.>300 ℃),具有低毒性、无致癌作用和廉价等优点,且三甲胺是唯一副产物。
该步反应为SN2反应,因此碱在吡啶酚的离子化过程中起关键作用,本文在DMF为溶剂,反应温度为145 ℃,n(碱) ∶n(4) ∶n(四甲基氯化铵)=1.0 ∶1.0 ∶1.1条件下考察了碱对甲基化反应的影响,结果见表3。由表3可见,碱性越强,离子化效果越好,4的收率越高,考虑到成本及强碱对设备要求高等因素,本文选择了KOCH3作为反应用碱。
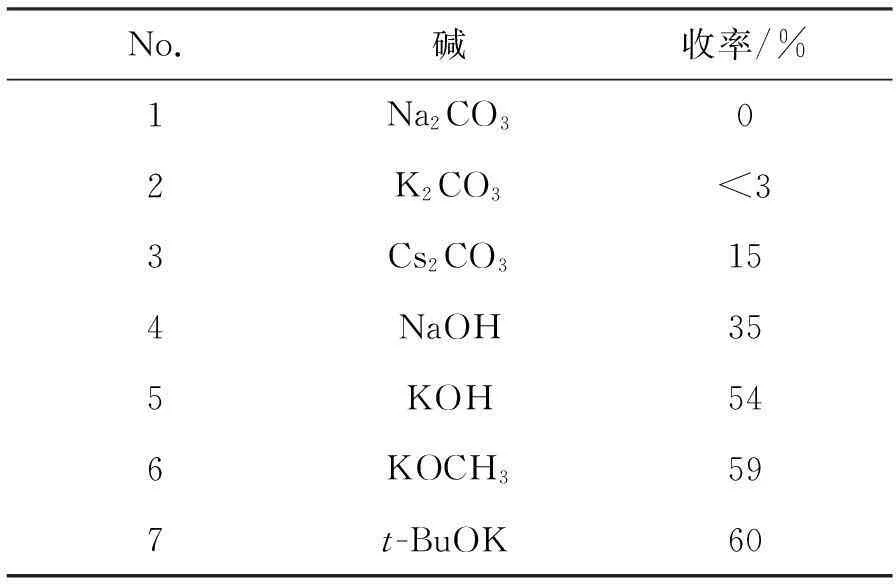
表3 碱对4收率的影响
同时,本文在145 ℃,n(KOCH3) ∶n(3) ∶n(四甲基氯化铵)=1.0 ∶1.0 ∶1.1条件下考察了溶剂对反应的影响,结果见表4。由表4可见,极性质子溶剂对该反应不利,极性非质子溶剂较非极性非质子溶剂利于反应的进行,干燥的DMSO为最佳溶剂选择,收率可达75%。最佳反应条件为:DMSO为溶剂,反应温度170 ℃,n(碱) ∶n(3) ∶n(四甲基氯化铵)=1.0 ∶1.0 ∶1.1,收率87%。
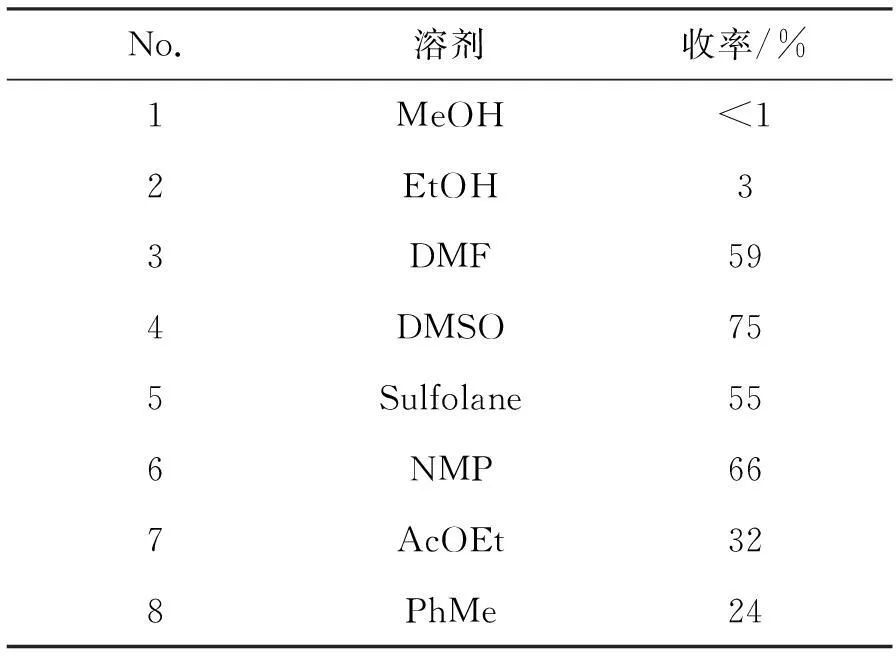
表4 溶剂对4收率的影响
2.3 5的合成工艺优化
在5的合成中,去质子化试剂的选择是该步反应的重点。本研究考察了多种常见的去质子化试剂,结果见表5。由表5可看出,LDA的去质子化效果最佳,但由于其成本高、反应温度要求低及设备要求高等原因,该研究最终选用正丁基锂作为去质子化试剂。

表5 去质子化试剂对5收率的影响
2.4 6和1的合成工艺优化
在1的合成中,文献[12]报道采用对5直接进行催化氢化的方法。该方法在直接还原过程中不可避免地有酮羰基还原副产物产生。本文以文献方法[13]为基础,采用先保护羰基制得6, 6经还原合成1。经改进,以甲苯替代毒性强的苯作为溶剂,对甲苯磺酸替代一水合对甲苯磺酸,并以6 eq.乙二醇作为酮羰基保护剂,以高达96%的收率制得6。
在1的合成中,文献[12-13]方法均以Rh/Al2O3为催化剂。本文考察了Pd/C, Rh/Al2O3和Rh/C三种催化体系。结果表明:Rh/C催化效果最好,当甲醇为溶剂,m(6) ∶m(Rh/C)=2 ∶1 ,n(6) ∶n(AcOH)=1.0 ∶1.1,反应温度70 ℃,氢气压力0.5 MPa时,6转化率为100%,水解后收率为97%。
3结论
以Donald小组[12]和沈昌茂等[13]的研究为基础,对以廉价易得的2-乙酰呋喃为起始原料的常山酮中间体的合成工艺进行了优化,总收率为40.2%,较文献[13]方法提高了32.9%,且放大实验显示优化后工艺条件稳定。与文献方法相比,该工艺具有收率高、反应清洁及成本低等优点。不足之处是运用n-BuLi作为去质子化试剂使得该步反应成本高,劳动保护强度大,总体上对工业生产仍有一定的指导意义。
参考文献
[1]Spira G, Mawasi N, Paizi M,etal. Halofuginone,a collagen type I inhibitor improves liver regeneration in cirrhotic rats[J].J Hepatol,2002,37(3):331-339.
[2]Tony L H, Guan Q N, Christopher Y C,etal. Halofuginone suppresses T cell proliferation by blocking proline uptake and inducing cell apoptosis[J].Int Immunopharmacology,2013,16(4):414-423.
[3]Giadinis N D, Papadopoulos E, Lafi S Q,etal. Efficacy of halofuginone lactate for the treatment and prevention of cryptosporidiosis in goat kids:An extensive field trial[J].Small Ruminant Research,2008,76(3):195-200.
[4]Herman J D, Rice D P, Ribacke U,etal. A Genomic and evolutionary approach reveals non-genetic drug resistance in malaria[J].Genome Biology,2014,15:1-14.
[5]Nelson E F, Craig W, Huang J M,etal. Halofuginone down-regulates Smad3 expression and inhibits the TGFbeta-induced expression of fibrotic markers in human corneal fibroblasts[J].Molecular Vision,2012,18:479-487.
[6]Broekaert N, Daeseleire E, Delezie E,etal. Can the use of coccidiostats in poultry breeding lead to residues in vegetables? an experimental study[J].J Agric Food Chem,2012,60(50):12411-12418.
[7]Wang Y H, Wang Z H, Jiang H Y,etal. Development of a monoclonal antibody-based enzyme-linked immunosorbent assay for the analysis of diclazuril in chicken tissues[J].Food Anal Methods,2013,6:1685-1692.
[8]杜会茹,张越,马东来,等. 抗球虫药常山酮的研究进展[J].黑龙江畜牧兽医,2013,2:29-31.
[9]Huong D T M, Giong V T, Van H T,etal. Synthesis of febrifuginol analogues and evaluation of their biological activities[J].Tetrahedron Letters,2014,55:7226-7228.
[10]张越,杜会茹,王永国,等.常山碱和异常山碱的合成研究进展[J].河北师范大学学报,2008,32(4):510-515.
[11]McLaughlin N P, Evens P, Pines M. The chemistry and biology of febrifugine and halofuginone[J].Bioorganic & Medicinal Chemistry,2014, 22:1993-2004.
[12]Donald F, Barringer J R, Berkelhammer G,etal. The stereochemistry of Febrifugine.I.The equilibrium between cis-and trans-(3-substituted 2-piperidyl)-2-propanones[J].J Org Chem,1973,38(10):1933-1937.
[13]沈昌茂,汪世新,王杰. 禽兔抗球虫药——常山酮合成研究Ⅱ.中间体(3-甲氧基-2-哌啶基)-丙酮的制备[J].江苏农学院报,1988,9(4):17-20.
[14]戴桂元,胡涛,王玉成. 2-甲基-3-羟基吡啶的合成研究[J].化学试剂,2001,23(6):370-371.
[15]周泽建,王京刚,刘世普,等. 2-乙基-6-甲基-3-羟基吡啶的合成优化[J].河北化工,2007,30(11):18-19.
[16]Maras N, Polanc S, Kocevar M. Microwave-assisted methylation of phenols with tetramethylammonium chloride in the presence of K2CO3or Cs2CO3[J].Tetrahedron,2008,64:11618-11624.

《合成化学》加入超星公司学术期刊“域出版”
为进一步方便读者,紧跟“互联网+”潮流,《合成化学》于2015年6月起正式加入超星公司学术期刊“域出版”。“域出版”是借助移动出版技术,通过“以智带栏”的模式,主要依托移动智能终端的在线学术交流互动平台。该平台的APP(支持Android和ios)将于7月底上线。
欢迎广大读者浏览下载。
《合成化学》编辑部
Process Improvement on the Synthesis of Halofuginone Intermediate
WANG Chao-jie,WAN Yang,SONG Wei-qiang,LU Qun*
(School of Life Science and Engineering, Southwest Jiaotong University, Chengdu 610031, China)
Abstract:Halofuginone intermediate, 1-(3-methoxypiperidin-2-yl)propan-2-one, was synthesized by ring-expansion, methylation, deprotonation, nucleophilic addition, ketalation, hydrogenation and hydrolysis, using 2-acetylfuran as the starting material. The overall yield was 40.2%. The structure was confirmed by1H NMR and ESI-MS.
Keywords:2-acetylfuran; halofuginone; intermediate; 1-(3-methoxypiperidin-2-yl)propan-2-one; synthesis; process improvement
中图分类号:O621.3
文献标志码:A
DOI:10.15952/j.cnki.cjsc.1005-1511.2016.01.15016
作者简介:王超杰(1988-),男,汉族,河南尉氏人,硕士研究生,主要从事有机合成研究。 E-mail: wangchaojie1130@163.com
基金项目:四川汪氏动物保健有限公司资助项目
收稿日期:2015-01-14;
修订日期:2015-10-15
猜你喜欢
高等学校化学学报(2022年4期)2022-06-10
小学生优秀作文(低年级)(2022年3期)2022-03-29
中国现代中药(2022年12期)2022-02-18
传媒评论(2019年4期)2019-07-13
铜仁学院学报(2018年6期)2018-07-05
中成药(2017年4期)2017-05-17
中成药(2017年3期)2017-05-17
西安文理学院学报(自然科学版)(2016年4期)2016-12-19
中国烟草学报(2016年3期)2016-11-23
质谱学报(2015年5期)2015-03-01
