
17α,21-二羟基孕甾-4-烯-3,20-二酮-17-戊酸酯的合成
2016-02-25 05:47吴庆安吴红卫
合成化学 2016年1期
吴庆安, 吴红卫, 柏 挺
(1. 浙江工业大学 化学工程学院,浙江 杭州 310014; 2. 上海新华联制药有限公司,上海 201400)
·制药技术·

通信联系人: 吴红卫,硕士研究生, E-mail: heyc27@sina.cn
17α,21-二羟基孕甾-4-烯-3,20-二酮-17-戊酸酯的合成
吴庆安1,2, 吴红卫1*, 柏挺2
(1. 浙江工业大学 化学工程学院,浙江 杭州310014; 2. 上海新华联制药有限公司,上海201400)
摘要:以雄烯二酮为起始原料,经加成、硅醚化、环加成-质子脱硅基化及置换等7步反应合成了氢化可的松的中间体——17α,21-二羟基孕甾-4-烯-3,20-二酮-17-戊酸酯,总收率75%,纯度>96%,其结构经1H NMR和ESI-MS确证。
关键词:雄烯二酮; 氢化可的松中间体; 17α,21-二羟基孕甾-4-烯-3, 20-二酮-17-戊酸酯; 合成
氢化可的松,化学名为11β,17α,2l-三羟基孕甾-4-烯-3,20-二酮,是肾上腺糖皮质激素药物,在激素类药物中占有重要地位。氢化可的松能够影响糖代谢,具有抗病毒、消炎、抗过敏及抗休克等作用[1-4]。其传统合成方法存在的主要问题有:(1)薯蓣资源的日渐枯竭;(2)铬废水的处理;(3)碘化反应成本高;(4)利用微生物氧化法引入11β-OH的选择性较低。
鉴于薯蓣资源的紧缺及传统合成路线的缺陷,本文通过对多种皮质激素甾体侧链引入方法的研究比较[5-12],借鉴了Livingston的合成理念,以植物甾醇发酵物雄烯二酮(4-AD)为起始原料,经加成、硅醚化、环加成-质子脱硅基化及置换等7步反应合成了氢化可的松关键中间体——17α,21-二羟基孕甾-4-烯-3,20-二酮-17-戊酸酯(1, Scheme 1),总收率75%,纯度>96%,其结构经1H NMR和ESI-MS确证。
1实验部分
1.1 仪器与试剂
YRT-3型熔点仪(温度未校正);Anance Ⅲ-500MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);Decax-60000 LCQ Deca XP型液相色谱-离子阱质谱联用仪;Agilent 1100型高效液相色谱仪。
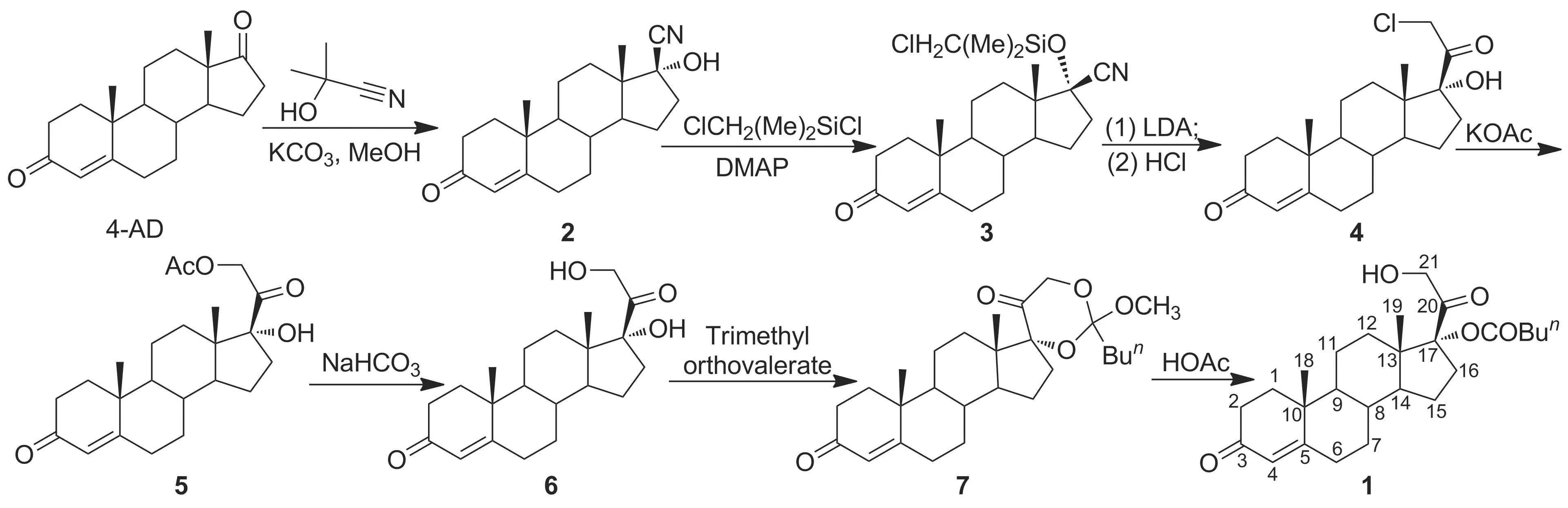
Scheme 1
4-AD,纯度98%,上海新华联制药有限公司;其余所用试剂均为分析纯,上海迈瑞尔化学技术有限公司。
1.2 合成
(1) 17β-氰基-17α-羟基雄甾-4-烯-3-酮(2)的合成[13-15]
氮气保护下,在反应瓶中依次加入4-AD 5.01 g(17.49 mmol)和甲醇10 mL,搅拌使其溶解;加入丙酮氰醇2.50 mL(27.38 mmol),缓慢滴加K2CO30.45 g的水(10 mL)溶液,滴毕,于30 ℃反应25 h。冷却至0 ℃,加入冰水10 mL,过滤,滤饼用饱和食盐水洗至中性,用甲醇重结晶,干燥得白色粉末2 5.28 g,收率96.30%,纯度99.07%(HPLC,下同), m.p.158.6~159.3 ℃(159.0~160.5 ℃[16]);1H NMR(DMSO-d6)δ:0.87(s, 3H, 18-H), 1.16(s, 3H, 19-H), 2.41(m, 2H, 2-H), 5.63(s, 1H, 4-H); ESI-MSm/z: 314.2{[M+H]+}。
(2) 17β-氰基-17α-羟基雄甾-4-烯-3-酮-17-(二甲基氯甲基)硅醚(3)的合成[12]
在反应瓶中依次加入2 5.18 g(16.53 mmol), 4-二甲氨基吡啶(DMAP)0.10 g,二氯甲烷75 mL和Et3N 4.78 mL,冷却至0 ℃,加入氯甲基二甲基氯硅烷3.20 mL(24.29 mmol),于5~10 ℃反应2 h。冷却至0 ℃,加入冰水15 mL淬灭反应,用10%盐酸调至pH 3,静置分层,水相用二氯甲烷(2×15 mL)萃取,合并有机相和萃取液,依次用饱和NaHCO3溶液和饱和NaCl溶液洗涤,无水硫酸钠干燥,减压浓缩,干燥得米白色固体3 6.85 g,收率98.67%,纯度99.07%, m.p.143.7~145.9 ℃(143~146 ℃)[17];1H NMRδ: 0.38(s, 6H, SiCH3), 0.97(s, 3H, 18-H), 1.22(s, 3H, 19-H), 2.88(m, 2H, 2-H), 5.75(s, 1H, 4-H); ESI-MSm/z: 420.2[M+]。
(3) 17α-羟基-21-氯孕甾-4-烯-3,20-二酮(4)的合成[12]
氮气保护下,将3 5.15 g(12.26 mmol)溶于无水THF(75 mL)中,冷却至-72 ℃,搅拌下缓慢滴加2 mol· L-1LDA的THF(15.33 mL)溶液,滴毕;于-40~-35 ℃反应4 h;于-20~-30 ℃缓慢滴加浓盐酸25 mL,滴毕,加入水60 mL,于室温反应12 h。于35 ℃减压蒸馏(析出土黄色固体),抽滤,滤饼用水洗涤,用二氯甲烷/乙醇(V/V=1/10)11 mL精制,干燥得淡黄色固体4 4.10 g,收率91.64%,纯度95.23%, m.p.228.2~230.4 ℃(234~235 ℃[18]);1H NMR(DMSO-d6)δ: 0.55(s, 3H, 18-H), 1.14(s, 3H, 19-H), 2.57(m, 2H, 2-H), 4.47(d,J=12.0 Hz, 1H, 21-CH), 4.77(d,J=9.0 Hz, 1H, 21-H), 5.55(s, 1H, 17-OH), 5.64(s, 1H, 4-H); ESI-MSm/z: 365.2[M+]。
(4) 17α,21-二羟基-孕甾-4-烯-3,20-二酮-21-乙酸酯(5)的合成[19-20]
将4 6.95 g (19.05 mmol)溶于DMSO(70 mL)中,搅拌下加入KOAc 17.38 g(177.09 mmol),缓慢升温至60 ℃,保温反应3 h。冷却至室温,缓慢倒入1.5 L冰水中(析出沉淀),静置4~6 h,过滤,滤饼用水洗涤,用乙醇14 mL精制,干燥得白色粉末5 7.14 g,收率96.50%,纯度98.29%, m.p. 223.3~226.4 ℃(225~226 ℃[11]);1H NMRδ: 0.73(s, 3H, 18-H), 1.20(s, 3H, 19-H), 2.19(s, 3H, OCH3), 4.87(d,J=9.0 Hz, 1H, 21-H), 5.09(d,J=9.0 Hz, 1H, 21-H), 5.75(s, 1H, 4-H); ESI-MSm/z: 389.2{[M+H]+}。
(5) 17α, 21-二羟基-孕甾-4-烯-3, 20-二酮(6)的合成[19-20]
将5 5.06 g(13.03 mmol)溶于甲醇(250 mL)中,搅拌下升温至50 ℃,加入饱和碳酸氢钠溶液25 mL,保温反应2 h。用2%盐酸调pH至7,于55 ℃减压浓缩至无溶剂蒸出,加入水100 mL,搅拌2 h,于冰箱中静置1 h,过滤,滤饼用乙酸乙酯重结晶,干燥得乳白色固体6 4.08 g,收率90.41%,纯度97.58%, m.p.202.1~204.4 ℃(207~208 ℃[21]);1H NMRδ: 0.68(s, 3H, 18-H), 1.16(s, 3H, 19-H), 2.66(s, 1H, 17-OH), 3.13(s, 1H, 21-OH), 4.28(d,J=15.0 Hz, 1H, 21-H), 4.64(d,J=15.0 Hz, 1H, 21-H), 5.71(s, 1H, 4-H); ESI-MSm/z: 347.4{[M+H]+}。
(6) 1的合成[22]
在反应瓶中依次加入THF 50 mL, 6 5.05 g(14.58 mmol)和对甲苯磺酸0.15 g(0.87 mmol),氮气保护,搅拌下升温至60 ℃,加入原戊酸三甲酯4.80 mL(26.69 mmol),保温反应3 h。冷却至-10 ℃,滴加冰醋酸/水(V/V=3/1)混合溶液20 mL,滴毕;于0 ℃反应12 h。用2%氢氧化钠溶液调至pH≈7,加水50 mL,于80 ℃常压蒸馏至无液体蒸出,冷却,静置,抽滤,滤饼用水洗涤,干燥得白色片状固体1 6.16 g,收率98.15%,纯度96.76%, m.p.118.7~121.9 ℃(115~118 ℃[22]);1H NMRδ: 0.69(s, 3H, 18-H), 0.90(t,J=6.0 Hz, 3H, CH3in Bu), 1.16(s, 3H, 19-H), 2.41(s, 1H, 21-OH), 4.81(d,J=15.0 Hz, 1H, 21-H), 5.07(d,J=15.0 Hz, 1H, 21-H), 5.71(s, H, 14-H); ESI-MSm/z: 431.4{[M+H]+}。
2结果与讨论
在2的合成中,用丙酮氰醇代替文献[15]方法中用到的剧毒品氰化钾时,反应的立体选择性不高。但当反应体系中含有钾离子时,立体选择性有明显提高,收率也较高(96.32%),纯度99.07%,异构体含量<1%。
在3的合成中,以氯甲基二甲基氯硅烷为硅醚化试剂,不需要对C-3位羰基进行保护,即可在强碱作用下发生分子内的环加成反应。随后,3在酸性条件下水解生成4。该反应不需要引入碘再置换,即可在C-21位引入一个氯原子。
在5的合成中,传统方法中C-21位碘被AcO-置换时[19-20],需要加入冰醋酸与I-形成离子氛,从而阻止I-的亲核性,促进反应的进行。而由4合成5时,因Cl-亲核性比I-小,所以不需要加入冰醋酸,反应就能顺利进行。如加入冰醋酸反而会阻碍反应的发生,使反应时间大大延长,杂质增大。
在6的合成中,水解所用碱的选择很重要,当所用碱的碱性较强(如氢氧化钠、氢氧化钾、碳酸钾、碳酸钠)时,反应中产生的杂质较多,而选用降温或降低碱浓度的方法均不能降低杂质的含量。当选用饱和碳酸氢钠为碱时,则能够很好的解决这一问题。
在1的合成中, 7水解生成17-单酯,主要副产物为17α,21-二羟基-孕甾-4-烯-3,20-二酮-21-戊酸酯。当采用冰醋酸代替无机强酸进行水解时,则大大提高了水解的选择性(>96%)。原因可能是C-17位的酯基与C-21位酯基相比,较易水解。当酸性较强时,主要生成副产物21-位单酯。
3结论
以植物甾醇发酵物雄烯二酮为起始原料,经加成、硅醚化、环加成-质子脱硅基化及置换等7步反应合成了17α,21-二羟基孕甾-4-烯-3,20-二酮-17-戊酸酯(1),总收率75%,纯度96%。1可通过微生物氧化、水解制备氢化可的松。
该合成路线与传统的由薯蓣皂素为原料制备氢化可的松的合成路线相比具有以下优点:原料廉价易得,储量丰富;引入侧链合理,C-3的羰基无需加以保护;缩短了合成路线,并通过对影响因素的研究确定了反应的最优条件;整个合成过程中没有用到重金属,符合绿色环保的要求;没有经过21-H的碘代反应,降低了生产成本,适合工业化生产。
参考文献
[1]Neeck G. Fifty years of experience with cortisone therapy in the study and treatment of rheumatoid arthritis[J].Ann N Y Acad Sci,2002,966:28-38.
[2]Sakuma E, Soji T, Herbert D C. Effects of hydrocortisone on the formation of gap junctions and the abnormal growth of cilia within the rat anterior pituitary gland[J].Anatomical Record,2001,262(2):169-175.
[3]Florio S, Ciarcia R, Crispino L,etal. Hydrocortisone has a protective effect on cyelosporineA-induced cardiotoxieity[J].J Cell Phys,2003,195(1): 2l-26.
[4]Gloor B, Uhl W, Tcholakov O,etal. Hydrocortisone treatment of early SIRS in acute experimental pancreatitis[J].Dig Dis Sci,2001,46(10):2154-2161.
[5]Murahashi S, Saito T, Hanaoka H,etal. Ruthenium-catalyzed oxidative transformation of alkenes to alpha-ketols with peracetic acid. Simple synthesis of cortisone acetate[J].J Org Chem,1993,58(11):2929-2930.
[6]Nitta I, Fujimori S, Haruyama T,etal. The synthesis of the corticoid side chain.Ⅲ.a new synthesis of 17α,21-dihydroxypregna-1,4-diene-3,20-dione-17,21-diacetate from androsta-1,4-diene-3,17-dione[J].Bull Chem Soc Jpn,1985,58(3): 981-986.
[7]Carruthers N I, Garshasb S, McPhail A T. Synthesis of corticoids from 9.alpha-hydroxyandrost-4-ene-3,17-dione[J].J Org Chem, 1992,57(3):961-965.
[8]Barton D H R, Motherwell W B, Zard S Z. A simple construction of the hydroxyl-ketone side chain of corticosteroids from 17-oxo-steroids via nitro-olefins[J].J Chem Soc Chem Commun,1982,10:551-552.
[9]Daniewshi A R, Wojciechowska W. An improved synthesis of the corticoid side chain[J].Synthesis,1984,2:132-133.
[10]Hanna I, Fetizon M, Goulaouic P. 1,4-Dioxene(2,3-dihydro-1,4-dioxine)in organic synthesis.Part 9.preparation of biologically active side-chains from 17-oxosteroids[J]. J Chem Soc Perkin. Trans I,1990,15(4):1107-1110.
[11]何明华, 廖清江. 氢化可的松中间体孕甾-4-烯-17α,21-二醇-3,20-二酮-21-醋酸酯的合成[J].Chinese Journal of New Drugs,2010, 19(3):233-235.
[12]Livingston D A, Petre J E, Bergh C L. Intramolecular cyanohydrin elaboration. Construction of corticosteroids from 17-ketosteroids[J].J Am Chem Soc,1990,112:6449-6450.
[13]Schwede W, Fritzemeier K H, Halfbrodt W,etal. Synthesis and biological activity of 11,19-bridged progestins[J].Steroids,1998,63:166-177.
[14]Miyashita M, Hara S, Yoshikoshi A. Regiospecific synthesis ofb-Thujaplicin(Hinokitiol) from 2-isopropylphenol[J].J Org Chem,1987,52:2602-2604.
[15]Nitta I, Fujimori S, Ueno H. The synthesis of the corticoid side chain.Ⅰ.An improved method for the preparation of 17α-hydroxyprogesterone form androst-4-ene-3,17-dione[J].Bull Chem Soc Jpn,1985,58(3):978-980.
[16]VanRheenen V H. Cyanohydrin transformation of steroids:US 4 500 461[P].1985.
[17]Livingston D A, Pearlman B A, Denmark S,etal. 17β-cyano-9α,17α-dihydroxyandrost-4-en-3-one:US 4 921 638[P].1990.
[18]Borrevang P. 21-Derivatives of compound S,cortisone,and hydrocortisone[J].Acta Chem Scand,1955,9(4):587-594.
[19]卢彦昌, 丁丽. 氢化可的松及其衍生物的制备:CN 101 397 321[P].2009.
[20]方伟明, 唐苏杭. 一种氢化可的松的制备方法:CN 102 367 262[P].2012.
[21]Julian P L, Meyer E W, Karpe W J,etal. Sterols. XI.l7α-Hydroxy-11-desoxycorticosterone[J].J Am Chem Soc,1950,72(11):5145-5147.
[22]Ercoli A, Gardi R, Brinaza C,etal. 17-Monoesters of 17α,21-dihydroxy steroids:BE 619 180[P].1962.
Synthesis of 17α,21-Dihydroxypregn-4-ene-3,20-dione-17-valerate
WU Qing-an1,2,WU Hong-wei1*,BAI Ting2
(1. College of Chemical Engineering, Zhejiang University of Technology, Hangzhou 310014, China;
2. Shanghai New Hualian Pharmaceutical Company Limited, Shanghai 201400, China)
Abstract:The intermediate of hydrocortisone, 17α,21-dihydroxypregn-4-ene-3,20-dione-17-valerate, was synthesized by a seven-step reaction of addition, silicon etherification, cycloaddition-protiodesilylation and so on, using androstendione as the starting material. The total yield was about 75% and purity was more than 96%. The structure was confirmed by1H NMR and ESI-MS.
Keywords:androstendione; hydrocortisone intermediate; 17α,21-dihydroxypregn-4-ene-3,20-dione-17-valerate; synthesis
中图分类号:O629.8; O621.3
文献标志码:A
DOI:10.15952/j.cnki.cjsc.1005-1511.2016.01.15009
作者简介:吴庆安(1962-),男,汉族,湖南新田人,博士,副研究员,主要从事有机合成和天然生物活性物质的化学修饰研究。 E-mail: wuqa@nhl-pharm.com
基金项目:上海市2014年度“科技创新行动计划”生物医药领域产学研医合作项目(14DZ1930100)
收稿日期:2015-01-07;
修订日期:2015-11-16
猜你喜欢
——非均布滤饼的局部比阻与平均比阻的测定与计算方法
化工装备技术(2020年4期)2020-09-09
流体机械(2020年5期)2020-06-24
广州化工(2020年11期)2020-03-07
吉林农业科技学院学报(2018年4期)2019-01-11
中国矿业(2017年2期)2017-02-28
科技传播(2016年17期)2016-10-10
西北师范大学学报(自然科学版)(2015年6期)2016-01-19
橡胶工业(2015年2期)2015-07-29
中国塑料(2014年12期)2014-10-17
西南军医(2014年5期)2014-04-25
