
分子遗传学检查诊断Crigler-Najjar综合征Ⅱ型3例*
2016-03-24 01:47史萍沈鉴东刘瑶邵宇云严友德金柯刘源李军蒋龙凤
实用肝脏病杂志 2016年6期
史萍,沈鉴东,刘瑶,邵宇云,严友德,金柯,刘源,李军,蒋龙凤
·遗传性肝病·
分子遗传学检查诊断Crigler-Najjar综合征Ⅱ型3例*
史萍,沈鉴东,刘瑶,邵宇云,严友德,金柯,刘源,李军,蒋龙凤
目的探讨采用分子遗传学检查诊断Crigler-Najjar综合征Ⅱ型的方法。方法在本科收治的3例高间接胆红素血症患者,抽取外周静脉血,提取基因组DNA,应用PCR法扩增尿苷二磷酸葡萄糖醛酸转移酶1A1基因(UGT1A1)所含5个外显子及其侧翼序列,进行DNA直接测序。结果3例患者均检出UGT1A1基因5号外显子存在c.1456 T>G(p.Y486D)纯合突变;Y486D位于第5外显子上,使1456位胸腺嘧啶(T)突变为鸟嘌呤(G),导致486位氨基酸由酪氨酸(Tyr)变为天冬氨酸(Asp)。结论当临床上高度怀疑Crigler-Najjar综合征Ⅱ型时,应尽早行分子遗传学检查,确定其基因突变位点,以明确诊断。
Crigler-Najjar综合征;尿苷二磷酸葡萄糖醛酸转移酶;基因突变
Crigler-Najjar综合征(CNS)属于遗传性非溶血性高间接胆红素血症[1],分为Ⅰ型和Ⅱ型。CN-Ⅰ型由Crigler和Najjar在1952年首次报道[2],患者尿苷二磷酸葡萄糖醛酸转移酶(UGT)活性完全缺乏,血清胆红素水平可高达340~850μmol/L[3],常死于胆红素脑病,或遗留有严重的神经系统后遗症。CN-Ⅱ型患者UGT活性部分缺乏,血清胆红素水平为85~340μmol/L,可被苯巴比妥诱导,终生使用苯巴比妥治疗可长期生存。已有研究证实,编码UGT的GUT1A1基因是CNS的遗传学基础[4],进行UGT1A1基因检测以及结合临床诊断已成为目前诊断CNS的主要方法。本文报道3例黄疸待查患者,结合实验室、肝脏穿刺组织病理学和分子遗传学检查诊断的CN-Ⅱ型患者,以供临床医生学习、借鉴。
1 资料与方法
1.1 病史特点2013年8~11月我科收治的3例男性患者,诊断时年龄分别为15岁、23岁和29岁。临床均有反复皮肤、眼黄,尿黄,乏力症状,皮肤黄染可于劳累后出现,病程分别为4年、3月和20年。前2例患者血清总胆红素(TBIL)分别为109.6 μmol/L和193.5μmol/L,间接胆红素(IBIL)分别为92.3μmol/L和185.4μmol/L,其余生化指标如丙氨酸氨基转移酶(ALT)、天门冬氨酸氨基转移酶(AST)、谷氨酰基转肽酶(GGT)、碱性磷酸酶(ALP)等均无异常。血常规、病毒性肝炎相关标志物、铜蓝蛋白、眼底检查、肝胆胰脾B超等检查无异常发现,无特殊用药史,无遗传性疾病家族史。第3例患者血清TBIL 266.6μmol/L,IBIL200.6μmol/L,ALT 734 U/L,AST 204 U/L,GGT 1180 U/L,外院肝胆胰脾B超检查提示胆囊壁粗糙,轻度脂肪肝,其余辅助检查无明显异常。予以保肝等治疗后转氨酶降至正常,但胆红素下降不明显。
1.2 肝活检第2和第3例患者进行了在B超引导下肝脏穿刺术检查。
1.3 UGT1A1基因检查患者及其父母签署知情同意书后,分别抽取外周静脉血2 m l,采用商品化基因组DNA提取试剂盒(天根,北京)抽提样本基因组DNA。采用Primer Premier 5.0设计UGT1A1基因外显子编码区及其侧翼序列扩增引物(表1),提交上海生工公司合成。采用Sanger法对PCR扩增产物测序,将测序结果与UGT1A1基因参比序列(NM-000463)进行比对分析。

表1 UGT1A1基因外显子编码区及其侧翼序列扩增引物
2 结果
2.1 临床特征分析本组3例患者血清TBIL在109.6~266.6μmol/L之间,以间接胆红素升高为主,予以护肝药物治疗后,血清转氨酶可降至正常,无溶血表现,无特殊用药史,血清铜蓝蛋白和眼底检查未见明显异常。第3例患者存在非酒精性脂肪肝。
2.2 肝组织学表现第2例患者肝脏穿刺组织病理学检查提示,肝细胞中度水肿变性,汇管区少许慢性炎细胞浸润;第3例患者肝脏小叶内肝细胞单板排列,肝细胞轻度水肿,少量肝细胞内可见脂褐素样色素沉积(图1)。

图1 肝组织学表现(HE,100×)
2.3 UGT1A1基因检测情况3例患者均检出UGT1A1基因5号外显子存在c.1456 T>G(p. Y486D)纯合突变。Y486D位于第5外显子上,使1456位胸腺嘧啶(T)突变为鸟嘌呤(G),导致486位氨基酸由酪氨酸(Tyr)变为天冬氨酸(Asp,图2)。
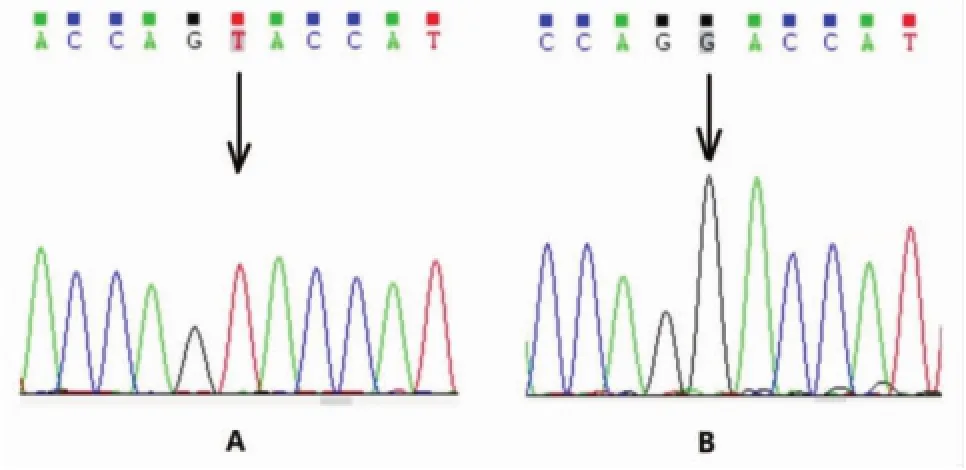
图2 UGT1A1基因参比序列(NM_000463)与患者UGT1A1基因测序结果比较
3 讨论
CNS是UGT1A1基因突变使其酶活性完全或部分丧失而导致的遗传性胆红素代谢障碍性疾病,分为Ⅰ型和Ⅱ型,发病率为百万分之一[5]。
UGT存在于肝细胞微粒体中,是生物体内进行第Ⅱ相生物转化时最重要的酶之一[6],代谢大量的外源性毒性物质和内源性物质。根据核苷酸序列的相似性,可将人类的UGT分为四类:即UGT1、UGT2、UGT3和UGT8,其中最重要的是UGT1和UGT2家族[7]。UGT1基因位于2q37,UGT1家族包括9个同工酶(UGT1A1、UGT1A3、UGT1A4、UGT1A5、UGT1A6、UGT1A7、UGT1A8、UGT1A9和UGT1A10)和4个假基因(UGT1A2P、UGT1A11P、UGT1A12P和UGT1A13P)[8]。只有UGT1A1编码以胆红素为底物的UGT1(B-UGT)[9],因此UGT1A1基因突变可导致胆红素代谢异常,表现为遗传性高间接胆红素血症,如CNS和Gilbert综合征(GS)。
自1992年[10]以来,已发现UGT1A1基因存在约140个不同的致病突变或变异位点[4]。CN-Ⅰ型是最严重的表型,而CN-Ⅱ型则介于CN-Ⅰ型和GS之间。多项研究发现,CN-Ⅱ型相关的分子学基础主要是UGT1A1基因错义突变及其他类型突变,可见于第1~5外显子编码区[3,11-16],第5外显子的第1456位T>G(p.Y486D)即是突变位点之一。Y486D纯合突变首先在日本CN-Ⅱ患者发现[17],Yamamoto et al[18]对UGT1A1酶活性研究显示,单一纯合型Y486D的UGT1A1酶活性为正常野生型酶活性的(7.6± 0.5)%。虽然酶活性显著下降但并未消失,可被苯巴比妥诱导,这也是CNS-Ⅱ型患者可用苯巴比妥治疗的分子学基础。本组3例患者虽未进行苯巴比妥诱导试验,但是均检出c.1456 T>G(p.Y486D)纯合突变,再结合患者临床症状及TBIL水平,符合CN-Ⅱ型诊断。然而,既往报道中CN-Ⅱ型又以复合杂合突变较多[13,19],主要基因型为G71R和Y486D突变,以亚洲地区报道最多见[3]。Maruo et al[13]报道1例患有CN-Ⅱ型的3月龄患儿,为复合杂合突变,体外酶活性研究显示包含p.[G71R;Y486D]突变的UGT1A1酶活性是野生型的6.1%。
本文中第2和第3例患者肝脏穿刺病理学检查提示肝细胞轻或中度水肿,汇管区少许慢性炎细胞浸润,少量肝细胞内可见脂褐素样色素沉积。这与前人研究结果类似。孙艳玲等[20]分析了155例先天性胆红素代谢障碍性肝病的病理研究结果,其中包括GS 115例,CNS 5例。GS和CNS的组织学基本表现为肝组织内小叶结构均完整,中央区周围/区域肝细胞浆内可见较细的棕褐色色素颗粒沉积,并且可存在肝细胞水样变性、脂肪变性以及汇管区炎细胞浸润。因此,这2例肝脏病理检查也佐证了CNS的诊断。但是,单纯依靠肝脏病理检查,并不能完全区分出GS与CNS,故仍需进行分子遗传学检查。
目前,遗传性非溶血性高间接胆红素血症的报道仍然较少,临床医生对于此类疾病的诊断较困难,即使已行肝脏穿刺病理检查也并不一定能够明确诊断。对于高度怀疑此类疾病的患者,应尽早行分子遗传学检查,确定基因突变位点,明确诊断并指导治疗。
[1]Strassburg CP.Hyperbilirubinemia syndromes(Gilbert-Meulengracht,Crigler-Najjar,Dubin-Johnson,and Rotor syndrome).Best Pract Res Clin Gastroenterol,2010,24(5):555-571.
[2]Crigler JJ,Najjar VA.Congenital familial nonhemolytic jaundice with kernicterus.Pediatrics,1952,10(2):169-180.
[3]Huang CS,Tan N,Yang SS,et al.Crigler-Najjar syndrome type 2.J Formos Med Assoc,2006,105(11):950-953.
[4]Sneitz N,Bakker CT,de Knegt RJ,et al.Crigler-Najjar syndrome in the Netherlands:identification of four novel UGT1A1 alleles,genotype-phenotype correlation,and functional analysis of 10 missense mutants.Hum Mutat,2010,31(1):52-59.
[5]潘丽丽,石岩,阴怀清,等.Crigler—Najjar综合征Ⅱ型UGT1A1基因突变一例报道暨文献复习.中国新生儿科杂志,2013,28(3):180-183.
[6]郭栋,庞良芳,周宏灏.尿苷二磷酸葡萄糖醛酸基转移酶基因多态性的研究进展.生理科学进展,2010,41(2):107-111.
[7]Mackenzie PI,Bock KW,Burchell B,et al.Nomenclature update for the mammalian UDP glycosyltransferase(UGT)gene superfamily.Pharmacogenet Genomics,2005,15(10):677-685.
[8]Owens IS,Basu NK,Banerjee R.UDP-glucuronosyltransferases: gene structures of UGT1 and UGT2 families.Methods Enzymol,2005,400:1-22.
[9]Sugatani J.Function,genetic polymorphism,and transcriptional regulation of human UDP-glucuronosyltransferase(UGT)1A1. Drug Metab Pharmacokinet,2013,28(2):83-92.
[10]Ritter JK,Yeatman MT,Ferreira P,et al.Identification of a genetic alteration in the code for bilirubin UDP-glucuronosyltransferase in the UGT1 gene complex of a Crigler-Najjar type I patient.JClin Invest,1992,90(1):150-155.
[11]Kadakol A,Ghosh SS,Sappal BS,et al.Genetic lesions of bilirubin uridine-diphosphoglucuronate glucuronosyltransferase(UGT1A1)causing Crigler-Najjar and Gilbert sy ndromes:correlation of genotype to phenotype.Hum Mutat,2000,16(4):297-306.
[12]Servedio V,D'Apolito M,Maiorano N,et al.Spectrum of UGT1A1 mutations in Crigler-Najjar(CN)syndrome patients:identification of twelve novel alleles and genotype-phenotype correlation.Hum Mutat,2005,25(3):325.
[13]Maruo Y,Ozgenc F,Mimura Y,et al.Compound heterozygote of a novel missense mutation(p.K402T)and a double missense mutation(p.[G71R;Y486D])in type II Crigler-Najjar syndrome. J Pediatr Gastroenterol Nutr,2011,52(3):362-365.
[14]Iijima S,Ohzeki T,Maruo Y.Hereditary spherocytosis coexisting with UDP-glucuronosyltransferase deficiency highly suggestive of Crigler-Najjar syndrome type II.Yonsei Med J,2011,52(2): 369-372.
[15]Costa E,Vieira E,Martins M,et al.Analysis of the UDP-glucuronosyltransferase gene in Portuguese patients with a clinical diagnosis of Gilbert a nd Crigler-Najjar syndromes.Blood Cells Mol Dis,2006,36(1):91-97.
[16]Nair KM,Lohse P,Nampoothiri S.Crigler-Najjar syndrome type 2:Novel UGT1A1 mutation.Indian J Hum Genet,2012,18(2): 233-234.
[17]Aono S,Yamada Y,Keino H,et al.Identification of defect in the genes for bilirubin UDP-glucuronosyl-transferase in a patient with Crigler-Najjar syndrome type II.Biochem Biophys Res Commun,1993,197(3):1239-1244.
[18]Yamamoto K,Sato H,Fujiyama Y,et al.Contribution of two missense mutations(G71R and Y486D)of the bilirubin UDP glycosyltransferase(UGT1A1)gene to phenotypes of Gilbert's syndrome and Crigler-Najjar syndrome type II.Biochim Biophys Acta,1998,1406(3):267-273.
[19]Liu WL,Li F,He ZX,et al.Analysis of bilirubin UDP-glucuronosyltransferase gene mutations in an unusual Crigler-Najjar syndrome patient.Mol Med Rep,2012,6(3):667-669.
[20]孙艳玲,赵景民,辛绍杰,等.几种主要的先天性胆红素代谢障碍性肝病的临床及病理研究.传染病信息,2008,11(5): 287-290.
(收稿:2016-04-21)
(本文编辑:陈从新)
Mutation of uridine diphosphate glucuronosyltransferase in three patients with Crigler-Najjar syndrom e type II
Shi Ping,Shen Jiandong,Liu Yao,et al.
Department of Infectious Diseases,First Affiliated Hospital,Nanjing Medical University,Nanjing 210029,Jiangsu Province,China
Jiang Longfeng,E-mail:phoenix0929@126.com
Ob jec tive To investigate the diagnosis of patients with Crigler-Najjar syndrome type II(CNII).M ethods Three patients with sustained unconjugated hyperbilirubinemia were admitted to our Department of Infectious Diseases,First Affiliated Hospital,Nanjing Medical University.The blood genome DNA was extracted,and five exons and their flanking sequences of uridine diphosphate glucuronosyltransferase 1A1 gene(UGT1Al)were amplified by polymerase chain reaction and the products were directly sequenced.Results Substitution of thymine for guanine at loci 1456 in the 5th exon existed leading to the change of amino acid from tyrosine to aspartate at 486th position of the corresponding protein(c.1456T>G:p.Y486D).So the three patients were confirmed with CN-Ⅱ.Conclusions A given patient with sustained unconjugated hyperbilirubinemia should be applied early to the gene analysis for the diagnosis of Crigler-Najjar syndrome.
Crigler-Najjar syndrome;Uridine diphosphate glucuronosyltransferase;Gene mutation
10.3969/j.issn.1672-5069.2016.06.005
国家科技“艾滋病和病毒性肝炎等重大传染病防治”重大专项(编号:2013ZX10002005);江苏省卫生计生委医学创新团队与领军人才培养基金(编号:LJ201121);中国肝炎防治基金会-天晴肝病研究基金(编号:CFHPC20132071)
210029南京市南京医科大学第一附属医院感染病科(史萍,刘瑶,邵宇云,严友德,金柯,刘源,李军,蒋龙凤);生殖医学科(沈鉴东)
史萍,女,27岁,医学硕士,住院医师。E-mail: shiping0218@126.com
蒋龙凤,E-mail:phoenix0929@126.com
猜你喜欢
中国现代医生(2022年19期)2022-11-04
电子科技大学学报(2022年5期)2022-10-29
中国典型病例大全(2022年7期)2022-04-22
中国生殖健康(2020年4期)2021-01-18
福建基础教育研究(2019年10期)2019-05-28
中国生殖健康(2018年4期)2018-11-06
求学·理科版(2017年3期)2017-04-27
中国卫生标准管理(2015年16期)2016-01-20
湖北农业科学(2014年11期)2014-09-10
中国中医药现代远程教育(2014年23期)2014-03-01
