
香榧转录组测序及生物信息学基础分析
2016-03-31 07:07易官美包燕春1宁波城市职业技术学院浙江宁波31550江西省南昌市青山湖风景区管理处江西南昌330039
山东农业大学学报(自然科学版) 2016年1期
易官美,包燕春1.宁波城市职业技术学院,浙江宁波31550.江西省南昌市青山湖风景区管理处,江西南昌330039
香榧转录组测序及生物信息学基础分析
易官美1*,包燕春2
1.宁波城市职业技术学院,浙江宁波315502
2.江西省南昌市青山湖风景区管理处,江西南昌330039
摘要:香榧具有重要的经济价值,但其基因组信息相对匮乏,限制了其分子生物学和基因功能的研究。本文以不同组织的香榧作为研究对象,采用新一代高通量测序技术平台Illumina HiSeq™2000对香榧转录组进行测序和数据分析,共得到37,349,086个reads片段,总碱基数为4.35 G。利用组装软件,对获得的高质量序列进行组装,共得到104,636个Unigene,平均长度为784 nt,N50为1,702。将Unigene序列与公共数据库进行比对,28,766个Unigenes获得了注释。其中26,856个Unigene在NR蛋白数据库中获得注释,24,003个Unigenes在NT数据库中获得注释,21,401个Unigene在Swiss-Prot蛋白数据库中获得注释,16,137个Unigene在COG数据库中获得注释,11,410个Unigene在GO数据库中获得注释。根据KEGG注释信息,18,564个Unigene被划分到256个代谢途径中。SSR位点搜索发现,在4,217个Unigene中含有4,706个SSR位点。分析所获得的转录组数据,将为香榧功能基因的克隆,基因的表达,指纹图谱构建和分子标记辅助选育奠定基础。
关键词:香榧;转录组;高通量测序;SSR
香榧(Torreya grandis Fort. ex Lindl. cv. merrillii)为红豆杉科(Taxaceae)榧树属(Torreya)榧树(Torreya grandi Fort. ex Lindl.)的栽培变种。榧树起源于中国,在中国的湖南、江西、安徽、江苏、浙江和福建等省均有自然分布。榧树是世界上比较稀有的经济树种之一,既可以食用,也可以药用,还是绿化观赏的常用树种。香榧干果风味独特,富含营养,有止咳化痰、润肺明目、杀虫杀菌等多种功效;另外,香榧中含有的紫杉醇是一种具有抗癌活性的生理活性物质[1,2]。
利用基因组、转录组及蛋白质组学等各种组学技术可以揭示细胞生理活动的规律、探明生物代谢的机理[3]。转录组是某一物种或特定的细胞在某一个时间段或特定的环境条件下产生的所有转录物的集合。利用转录组学技术可以了解到基因的功能和基因的结构,可以揭示器官以及细胞在特定生物学过程中的分子机制[4]。近年来,转录组学的研究应用广泛,已应用于多种树木,主要有红豆杉(Taxus mairei)[5]、木榄(Bruguiera gymnorrhiza)[6]、刚毛柽柳(Tamarix hispida)[7]、桉树(Eucalyptus robusta)[8]、日本落叶松(Larix leptolepis)[9]、油桐(Vernicia fordii)[10]、橡胶树(Hevea brasiliensis)[11]和茶树(Camellia sinensis)[12]等。
目前,对香榧的研究主要集中在生理生化特性、栽培技术[1,2]和居群遗传学[13]等方面,缺少对其基因组学以及转录组学的研究。本研究利用Illumina高通量测序技术对香榧的转录组进行测序,利用生物学软件将获得的数据进行拼接组装,再利用生物信息学技术将获得的Unigene进行基因功能注释、基因功能分类以及代谢途径的分析,以期为香榧重要性状基因的克隆及基因功能的分析、分子标记的开发以及遗传图谱的构建奠定良好的基础。
1 材料和方法
1.1材料来源
香榧的叶片、幼果和枝干组织采集于浙江省宁波市奉化市锦溪村香榧种植基地(北纬29°32′24",东经121°11′50.8",海拔540 m)属亚热带季风性气候,四季分明,温和湿润,年均气温16.3℃,最高气温40.5℃,最低气温为-11.1℃。年平均降水量1500 mm。日照时数1850 h,无霜期232 d,生长于海拔500 m左右的山坡。采集8年生正常健壮的香榧嫁接苗,同一株香榧个体上的叶片,幼果,枝干组织各2 g,进行等量混样。立即用液氮速冻,在-80°C冰箱中冷冻保存备用。
1.2方法
1.2.1RNA提取总RNA的提取方法按照Invitrogen公司的Trizol Reagent说明书进行。DNA的消化处理按Promega公司DNaseⅠ的方法进行。RNA完整性检测利用Agilent的BioAnalyzer 2 100,样品RIN≥8。
1.2.2转录组测序及序列组装香榧cDNA文库的构建采用TruSeq RNA Sample Preparation Kit (Illumina)进行。利用带有Oligo(dT)的磁珠对mRNA进行富集。然后将打断成短片段的mRNA为模板,合成cDNA的第一条链。然后将cDNA第一条链与缓冲液、RNase H、dNTPs以及DNA Polymerase I配制成第二链合成的反应体系,合成cDNA第二条链。cDNA的纯化使用QiaQuick PCR试剂盒进行,然后纯化后的cDNA做末端修复。将修复后的cDNA 3’末端加上碱基“A”并连接上接头,随后对片段的大小进行选择。最后一步进行PCR反应,构建测序文库。利用Illumina HiSeq™2000对建好的测序文库进行测序。将测序获得的原始数据去除接头的序列、两端低质量的序列以及低度复杂的序列,数据的组装利用Trinity[14]软件进行。
1.2.3功能注释首先,通过Blastx程序将Unigene序列与蛋白数据库NR、Swiss-Prot(Evalue<0.00001)进行比对,并通过Blastn程序将Unigene与核酸数据库nt(Evalue<0.00001)进行比对,获得Unigene最高序列相似性的蛋白,便获得了该Unigene的蛋白质功能注释信息[14]。然后用Blastx将所得Unigene序列比对到COG(Clusters of Orthologous Groups of Proteins)数据库(Evalue<0.00001),获得COG数据库的功能注释及其功能分类。Gene Ontology(GO)注释信息使用软件Blast2GO[15]获得,然后使用WEGO软件[16]对Unigene进行GO功能的分类统计。根据KEGG的注释信息可进一步得到Unigene的Pathway途径注释。
1.2.4香榧SSR位点分析利用MISA软件(http://pgrc.ipk-gatersleben. de/misa/misa.html)进行SSR位点搜索,搜索参数设置为单、二、三、四、五和六碱基重复。
2 结果与分析
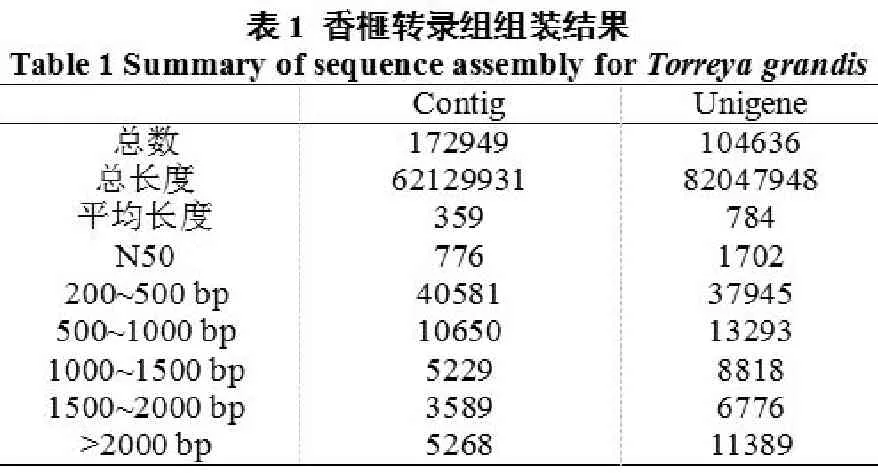
2.1转录组测序与组装
采用Illumina HiSeq™2000对香榧转录组进行测序,共得到reads片段37,349,086个,总碱基数约为4.35 G,GC的含量是42.31%,Q20(碱基测序错误率小于1%)是96.63%。这些结果说明,转录组测序质量良好,获得的数据可进一步分析。利用组装软件Trinity,对测序得到的高品质序列进行de novo从头组装,共获得了172,949个Contigs,平均的长度为359 nt,N50为776。其中,40,581个Contig长度为200~500 bp,占23.46%;10,650个Contig长度为500~1000 bp,占6.16%;14,086个Contig的长度为1,000bp以上,占8.14%(表1)。使用组装软件将Contig进一步的组装,获得了104,636个Unigene,平均长度为784 nt,N50为1,702。其中,37,945个Unigene长度为200~500 bp,占36.26%;13,293个Unigene长度500~1000 bp,占12.7%;8,818个Unigene长度为1000~1500 bp,占8.43%;6,776个Unigene长度为1000~1500 bp,占6.48%;2000 bp以上的Unigene有11,389个,占10.88%(表1)。N50长度可以用来衡量组装的完整度,Contig和Unigene的N50分别达到了776和1,702,表明了组装效果良好。
2.2Unigene的功能注释
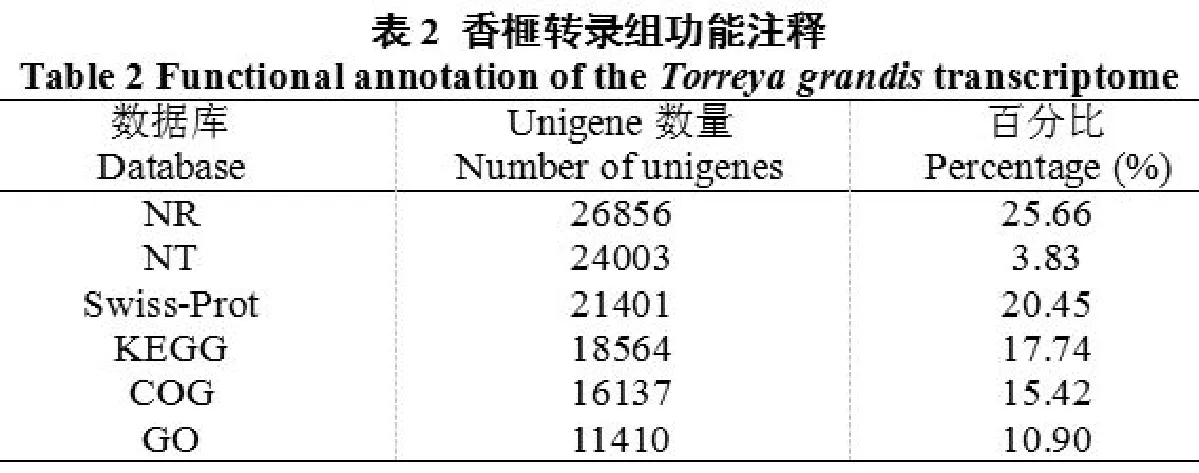
将Unigene序列通过Blast程序比对到数据库NR,NT,Swiss-Prot,KEGG,COG,GO (Evalue<0.00001),获得Unigene的功能注释。在上述数据库中获得注释的Unigenes有28,766个,其中在NR蛋白数据库中获得注释的Unigene有26,856个,在NT数据库中获得注释的Unigenes有24,003个,在Swiss-Prot蛋白数据库中获得注释的Unigene有21,401个(表2)。
2.3香榧转录组Unigene的COG功能分类
利用COG数据库可以对基因产物进行系统进化关系分类。本研究将获得的Unigene比对到COG数据库,16,137个Unigene获得了分类,根据功能可将这些Unigene分为25类(表3),涉及了大多数的生命活动。5,358个Unigene被归到一般功能预测类,占总数的33.20%。2,959个Unigene被归到翻译,核糖体结构及生物发生类,占总数的18.34%。2,654个Unigene被归到转录类,占总数的16.45%。2,106个Unigene被归到翻译后修饰,蛋白质折叠及分子伴侣类,占总数的13.05%。2,098个Unigene被归到信号传导机制类,占总数的13.00%。而被归到胞外结构类和核结构类的Unigene最少,分别为10个和3个。

表3 香榧转录组的COG分类Table 3 COG functional distribution of the Torreya grandis transcriptome
2.4香榧转录组Unigene的GO功能分类
Gene Ontology(GO)基因功能分类是国际上标准化的分类体系之一,生物体中基因与基因产物的属性由一套标准词汇表(Controlled vocabulary)来全面描。本研究中11,410个Unigene被归为55类(图1)。在参与的生物过程分类中归类为细胞过程(6,738个),代谢过程(5,684个),单一的生物过程(4,494个),生物调控(2,783个)中的Unigene最多。在所处的细胞位置分类中归类为细胞(5,260个),细胞器(3,921个),大分子复合物(1,851个),膜(1,388个)中的Unigene最多。在分子功能分类中参与催化活性(6,426个)、结合(5,310个)和转运活性(380个)的Unigene最多。
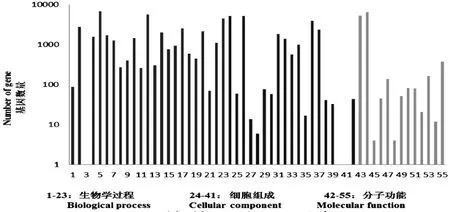
图1 香榧转录组的GO分类Fig.1 Gene ontology classification of the Torreya grandis transcriptome备注:生物学过程:1:生物附着2:生物调控3: cell killing细胞杀伤4:细胞成分和生物合成5:细胞过程6:发育过程7:定位系统建立8:生长;9:免疫系统过程10:定位11:移动12:代谢过程13:多机体过程14:多细胞组织过程15:生物过程的负调控16:生物过程的正调控17:生物过程的调控18:再生19:再生过程20:刺激应答21:律动过程22:信号传导23:单一的生物过程;细胞组成24:细胞25:细胞连接26:细胞部分27:细胞外基质28:胞外区要素29:胞外区30:胞外区部分31:大分子复合物32:膜33:膜要素34:膜附着腔35:核仁36:细胞器37:细胞器部分38:突触39:突触部分40:病毒体41:病毒体部分;分子功能42:抗氧化活性43:结合44:催化活性45:频道调节器活动46:电子载体活性47:酶调节活性48:通道活性的调节49:分子转导活性50:核酸结合的转录因子的活性51:蛋白结合转录因子活性52:刺激应答53:结构分子活性54:翻译调控因子活性55:转运活性.Note:Biological process:1: biological adhesion 2: biological regulation 3: cell killing 4: cellular component organization or biogenesis 5: cellular process 6: developmental process 7:establishment of localization 8: growth 9: immune system process 10: localization 11: locomotion 12: metabolic process 13: multi-organism process 14: multicellular organismal process 15: negative regulation of biological process 16: positive regulation of biological process 17: regulation of biological process 18: reproduction 19: reproductive process 20: response to stimulus 21: rhythmic process 22: signaling 23: single-organism process;Cellular component 24: cell 25: cell junction 26: cell part 27: extracellular matrix 28: extracellular matrix part 29: extracellular region 30: extracellular region part 31: macromolecular complex 32: membrane 33: membrane part 34: membrane-enclosed lumen 35: nucleoid 36: organelle 37: organelle part 38: synapse 39: synapse part 40: virion 41: virion part;Molecular function 42: antioxidant activity 43: binding 44: catalytic activity 45: channel regulator activity 46: electron carrier activity 47: enzyme regulator activity 48metallochaperone activity 49: molecular transducer activity 50: nucleic acid binding transcription factor activity 51: protein binding transcription factor activity 52: receptor activity 53: structural molecule activity 54: translation regulator activity 55: transporter activity
2.5香榧转录组Unigene的KEGG功能分类
利用KEGG数据库可以探索基因产物在细胞中的功能及所处的代谢通路,可以用来统计分析基因的产物在生物学上复杂的行为[17]。本次研究中18,564个Unigene被归到256个小类中。被归为代谢途径(Metabolicpathways)的Unigene数量最多,有3,123个,占16.82%。其次是剪接体(Spliceosome),有749个Unigene,占4.03%。第三大类是植物与病原物的互作,有723个Unigene,占3.89%(表4)。
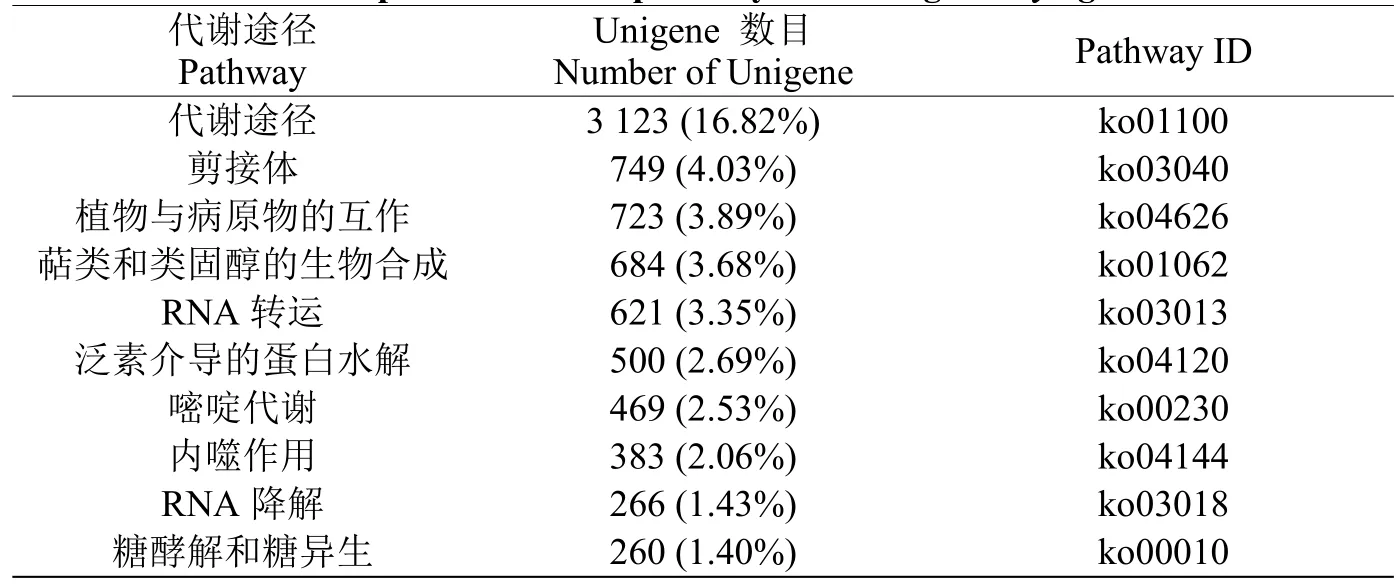
表4 Unigene数量最多的10个代谢通路Table 4 Top ten metabolic pathways involving Torreya grandis
2.6SSR分析
利用软件MISA搜索香榧Unigene的SSR位点,从104,636个Unigene检测到了4,706个SSR位点,出现的频率(检出的SSR个数和总Unigene的数目之比)为4.5%(表5)。在4,217个Unigene中分布着4,706 个SSR位点,其中414个Unigene含有1个及以上的SSR位点。SSR种类呈现多样化,单到六核苷酸的重复类型均检测到。其中位点数量多的SSR类型为三核苷酸,有1,677个;其次为二核苷酸,有1,139个;1,139个SSR位点为单核苷酸;四、五、六核苷酸SSR位点的数量相对较少,分别有69,257,614个。单核苷酸SSR的重复类型主要是A/T(945个);二核苷酸重复的主要类型是AT/AT(507个)、AG/CT (412个)、AC/GT(219个);三核苷酸重复主要类型是AAG/CTT(361个)、AGG/CCT(329个)、AGC/CTG(286个)、AAT/ATT(207个)。对这些SSR的鉴定,有助于开展香榧及其同属物种的遗传图谱构建及基因组差异分析研究。

表5 香榧SSR位点统计结果Table 5 Statistics of SSRs identified in Torreya grandis transcriptome
4 讨论
随着新一代高通量测序技术的不断进步,植物基因组的测序研究得到了快速的发展,但对香榧基因组的研究非常少。相对于全基因组序列的测定,转录组测序技术具有一定的优势,其一是从头装配转录本,不需要利用任何以前的基因序列信息;其次测序费用较低,普通的实验室也能承担。与其它测序技术相比,Illumina高通量测序技术获得的数据量大、测序速度快、效率比较高、成本相对较低,已在多种植物上得到应用[18]。本研究利用Illumina测序技术对香榧转录组进行了测序,共获得约4.4 G的原始数据,获得的数据量比较大,获得37,349,086个长度为90 bp的序列。利用生物学组装软件对这些90 bp的短序列进行了组装,共获得Unigene 104,636个。这些研究结果说明Illumina测序技术可以应用在香榧等基因组信息相对匮乏的物种上,并能有效得到转录组的信息。
将Unigene序列通过Blast程序与公共数据库Swiss-Prot、KEGG、NR、NT和COG进行比对,从中获得注释的Unigene有28,766个,其中在NR蛋白数据库中得到注释的Unigene有26,856个,在Swiss-Prot蛋白数据库中得到注释的Unigene有21,401个。104,636个Unigene中只有28,766个在上述的公共数据库中获得了注释信息。造成这样结果的原因有几种,一方面是有的Unigene的序列过短,而无法获得比对信息,另一方面有些Unigene可能是香榧特有的基因。本研究中获得的Unigene被归类到多个GO、COG以及KEGG子类中,说明这些组装的Unigene具有丰富的类型。
SSR分子标记操作相对简便、具有较好的重复性、获得的信息量高、覆盖广,已经应用在多个方面,包括植物分子辅助育种、遗传图谱的构建等方面[19-20]。本研究利用MISA软件分析了香榧转录组,获得了4,706个SSR位点。其中三核苷酸重复最多,其次是二核苷酸重复和单核苷酸重复,四、五、六核苷酸重复类型的数量较少。根据已有的报道,大多数植物的SSR类型主要以二核苷酸和三核苷酸为主,但是优势的重复单元也存在差异[21]。本次研究获得的优势重复单元是三核苷酸,其次是二核苷酸,这与葡萄(Vitis vinifera)[22]、大麦(Hordeum vulgare L.)[23]等植物一致,比松类高,低于经济树种茶。与其亲缘关系相近的红豆杉中以三核苷酸和六核苷酸为主,而且与模式植物拟南芥以三核苷酸为主也相近。水稻中的研究表明相对于三核苷酸和六核苷酸位于基因区而言,二核苷酸和四核苷酸主要位于非编码区,即基因间区域。从检出的频率来看,二核苷酸重复基元中出现最多的是AT/AT、其次是AG/CT和AC/GT,三核苷酸重复中的主要类型是AAG/CTT和AGG/CCT,这些重复基元在大多数双子叶植物中的出现频率较高[24]。香榧SSR重复基序的组成与双子叶植物更加接近,造成这种原因可能与密码子的偏倚性以及搜索条件的设置有关。由于香榧为非模式植物,可供参考的遗传信息相对比较少,因此对于其特异性的新基因的挖掘还有待进一步的研究。以本研究获得的转录组为基础,今后可以开展进一步的开发研究榧树的分子标记,从而对榧树的遗传结构和多样性展开研究,评估和保护其遗传资源;还可以克隆榧树中重要的生物活性成分的合成关键基因,以更好地利用其保健、药用价值和综合开发利用价值;也为功能基因的挖掘及优良性状的遗传改良等提供了大量的遗传数据资源[25]。
参考文献
[1]易官美,邱迎君.榧树的研究现状与展望[J].资源开发与市场,2013,29(8):844-847
[2]易官美,邱迎君,李晓花,等.榧树的地理分布与资源调查[J].安徽农业科学,2013,41(19):8200-8202
[3] Sun X,Zhou S,Meng F,et al. De novo assembly and characterization of the garlic(Allium sativum)bud transcriptome by Illumina sequencing[J]. Plant cell reports,2012,31:1823-1828
[4] Seungill K,Myung-Shin K,Yong-Min K,et al. Integrative structural annotation of de novo RNA-Seq provides an accurate reference gene set of the enormous genome of the onion(Allium cepa L.)[J]. DNARes,2015,22(1):19-27
[5] Da Cheng H,Guangbo G,Peigen X,et al. The first insight into the tissue specific taxus transcriptome via Illumina second generation sequencing[J]. PLoS one,2011,6(6):e21220
[6] Miyama M,Tada Y. Transcriptional and physiological study of the response of Burma mangrove(Bruguiera gymnorhiza)to salt and osmotic stress[J]. Plant molecular biology,2008,68(1-2):119-129
[7] Gao C,Wang Y,Liu G,et al. Expression profiling of salinity-alkali stress responses by large-scale expressed sequence tag analysis in Tamarix hispid[J]. Plant molecular biology,2008,66(3):245-258
[8] Mizrachi E,Hefer CA,Ranik M,et al. De novo assembled expressed gene catalog of a fast-growing Eucalyptus tree produced by Illumina mRNA-Seq[J]. BMC genomics,2010,11(6):681
[9] Zhang Y,Zhang S,Han S,et al. Transcriptome profiling and in silico analysis of somatic embryos in Japanese larch (Larix leptolepis)[J]. Plant cell reports,2012,31(9):1637-1657
[10]孙颖,谭晓风,罗敏,等.油桐花芽2个不同发育时期转录组分析[J].林业科学,2014,50(5):70-74
[11] Li D,Zhi D,Bi Q,et al. De novo assembly and characterization of bark transcriptome using Illumina sequencing and developmentof EST-SSRmarkersinrubbertree(Heveabrasiliensis Muell.Arg.)[J].BMCgenomics,2012,13(19):1-14
[12] Shi CY,Yang H,Wei CL,et al. Deep sequencing of the Camellia sinensis transcriptome revealed candidate genes for major metabolic pathways of tea-specific compounds[J]. BMC genomics,2011,12(1):1-19
[13]易官美,邱迎君.榧树居群遗传多样性的cpSSR分析[J].果树学报,2014,31(4):583-588
[14] Grabherr MG,Haas BJ,Yassour M,et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome[J]. Nature biotechnology,2011,29(7):644-652
[15] Conesa A,Götz S,García-Gómez JM,et al. Blast2GO: a universal tool for annotation,visualization and analysis in functional genomics research[J]. Bioinformatics,2005,21(18):3674-3676
[16] JiaY,LinF,HongkunZ,etal.WEGO:awebtoolforplottingGOannotations[J].Nucleicacidsresearch,2006,34(2):W293-W297
[17] Kanehisa M,Araki M,Goto S,et al. KEGG for linking genomes to life and the environment[J]. Nucleic acids research,2008,36(1):D480-D484
[18] Bie V,Clement L,Reumers J,et al. ViVaMBC: estimating viral sequence variation in complex populations from illumina deep-sequencing data using model-based clustering[J]. BMC bioinformatics,2015,16(1):1-11
[19] Agarwal M,Shrivastava N,Padh H. Advances in molecular marker techniques and their applications in plant sciences[J]. Plant cell reports,2008,27(4):617-631
[20]李炎林,杨星星,张家银,等.南方红豆杉转录组SSR挖掘及分子标记的研究[J].园艺学报,2014,4(4):735-745
[21] Wei W,Qi X,Wang L,et al. Characterization of the sesame(Sesamum indicum L.)global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers[J]. BMC genomics,2011,12:451
[22] Hong H,Jiang L,Ren Z,et al. Mining and validating grape(Vitis vinifera L.)ESTs to develop EST-SSR markers for genotyping and mapping[J]. Molecular Breeding,2011,28(2):241-254
[23] Varshney RK,Grosse I,Hähnel U,et al. Genetic mapping and BAC assignment of EST-derived SSR markers shows non-uniform distribution of genes in the barley genome[J]. Theoretical and Applied Genetics,2006,113(2):239-250
[24] Chagné D,Chaumeil P,Ramboer A,et al. Cross-species transferability and mapping of genomic and cDNA SSRs in pines[J]. Theoretical and Applied Genetics,2004,109(6):1204-1214
[25]邓楠,史胜青,常二梅,等.膜果麻黄种子不同发育时期的转录组测序分析[J].东北林业大学学报,2015,43(2):28-32
Sequencingand BioinformaticAnalysisfor Transcriptomeof Torreya grandis Fort.ex Lindl.cv.merrillii
YI Guan-mei1,BAO Yan-chun2
1. Ningbo City College of Vocational Technology, Ningbo 315502,China
2. Qingshanhu Landscape Adminstration Office of Nanchang City Jiangxi Province, Nanchang 330039,China
Abstract:Torreya grandis Fort. ex Lindl. cv. Merrillii is an economically important plant on both agriculture and ecology. However,the genomic information of this species has been less studied,leading to limited researching progresses in both molecular biology and gene functions. In the present study,we have sampled different tissues of T. grandi to conduct a transcriptomic analysis using the Illumina HiSeq™2000 technical platform. As a result,a total of 37,349,086 reads were obtained with the whole base number of 4.35 G. Based on the assembling of high quality reads,we identified a total of 104,636 Unigenes with an average length of 784 nt and N50 was 1,702 nt. Comparing these Unigene sequences with those in the public database,28,766 Unigenes were annotated in the Nr database,24,003 Unigenes were in the NT database,and 21,401 Unigenes were in the Swiss-Prot database. Moreover,based on the COG database and the GO database,we also found 16,137 Unigenes and 11,410 Unigenes were in both databases respectively. We further classified 18,564 Unigenes into 256 pathways according to the KEGG annotation information. Finally,we identified 4,706 SSR loci in 4,217 Unigenes via SSR loci searching. The obtained transcriptome data was thus as the first genomic-wide database serving for future studies of T. grandis in terms of functional gene cloning,gene expression,fingerprint construction and molecular marker-assisted breeding.
Keywords:Torreya grandis;transcriptome;Illumina sequencing;SSR
*通讯作者:Author for correspondence. E-mail:yiguanmei@nbcc.cn
作者简介:易官美(1968-),男,江西进贤人,硕士,副教授.主要从事植物资源学研究. E-mail:875013268@qq.com
基金项目:宁波市科技局农业重大专项项目(2014C11006);宁波市自然科学基金(2015A610267)
收稿日期:2015-05-13修回日期:2015-11-14
中图法分类号:Q37;S791.53
文献标识码:A
文章编号:1000-2324(2016)01-0019-06
猜你喜欢
恋爱婚姻家庭(2023年1期)2023-02-15
基层中医药(2018年8期)2018-11-10
浙江林业科技(2018年6期)2018-03-13
绿色中国(2017年19期)2017-05-25
中国中药杂志(2016年24期)2017-04-18
中国中药杂志(2017年4期)2017-03-28
中国中药杂志(2017年2期)2017-03-25
中国中药杂志(2017年3期)2017-03-20
科技创新导报(2016年28期)2017-03-14
中国中药杂志(2017年1期)2017-03-06
