
CCDC39基因突变致原发性纤毛运动障碍1例及其遗传咨询和产前诊断
2016-08-01 12:13祁媛媛王慧君张晓波王立波金婷婷钱莉玲
中国循证儿科杂志 2016年6期
祁媛媛 洪 达 王慧君 张晓波 王立波 金婷婷 钱莉玲
·论著·
CCDC39基因突变致原发性纤毛运动障碍1例及其遗传咨询和产前诊断
祁媛媛1洪 达1王慧君 张晓波 王立波 金婷婷 钱莉玲
目的 探讨对原发性纤毛运动障碍(PCD)患儿进行基因检测对指导优生优育的意义。方法 对1例临床诊断Kartagener综合征并随访4年的患儿进行家系全外显子组测序(WES),经高通量测序数据分析及临床诊断流程确定致病基因突变,根据基因诊断结果行针对性遗传咨询后对患儿母亲第2胎行羊水穿刺,分离羊水脱落细胞,提取基因组DNA并PCR 扩增羊水脱落细胞相应基因突变所在外显子,行直接Sanger测序。结果 临床随访发现患儿肺功能较诊断时明显下降,胸部CT支气管扩张较前加重。WES患儿中检测到CCDC39基因c.1819A>A/T, p.K607X无义突变和c.2447_2448het_delCA, p.T816Kfs*3移码突变,2个突变均为未报道的新突变,均经功能预测工具MutationTaster预测为致病突变,分析父母突变来源确定为复合杂合突变。同时结合患儿支气管黏膜活检电镜特征性的纤毛结构异常,认定该复合杂合突变为患儿的致病突变。患儿母亲第2 胎羊水细胞CCDC39基因2个突变位点所在外显子的Sanger测序结果显示,胎儿携带母亲来源的移码突变,无父亲的无义突变,为单杂合突变携带者。结论 采用WES检测有助于明确PCD患儿的遗传病因,在此基础上对患儿家庭行遗传咨询和产前基因诊断,可以指导优生优育。
原发性纤毛运动障碍; 全外显子组测序; 遗传咨询; 产前诊断
1 病例资料
1.1 临床资料 患儿,女,12岁,因“反复咳嗽1年余伴脓涕”于2012年10月首次至复旦大学附属儿科医院(我院)就诊。患儿系G1P1足月顺产,生后曾因“呼吸困难”转入我院新生儿科,X线胸片检查示肺炎,右位心。入院后予机械通气4 d后好转,改为鼻导管吸氧治疗1周后停氧,住院23 d好转出院。出院后患儿一般情况可,但易流脓涕,未予重视。入院前1年余,频繁呼吸道感染。入我院前1次门诊X线胸片示肺炎。入院查体:神志清楚,呼吸平,咽充血,两肺呼吸音粗,可闻及少许湿性啰音,心音有力,律齐,未闻及杂音,无杵状指。入院后肺部CT示,肺炎伴部分实变不张、支气管扩张。肺功能轻度限制性病变伴有小气道功能下降(表1)。心超显示镜像右位心,左位主动脉弓;腹部B超显示肝脾反位。患儿存在鼻窦炎、支气管扩张、合并内脏转位,考虑Kartagener综合征,遂行支气管镜黏膜活检,电镜检查示纤毛主杆排列紊乱,内动力蛋白臂与辐条缺失(图1)。确诊为原发性纤毛运动障碍(PCD)。

表1 PCD诊断时(8岁)与12岁随访时肺功能比较
注 VC:肺活量;FVC:用力肺活量;FEV1:1 s用力肺活量;FEV1/VC:1 秒率; FEF25%(50%、75%):呼出25%(50%、75%)肺活量时的呼气流速;MMEF:最大呼气中期流量
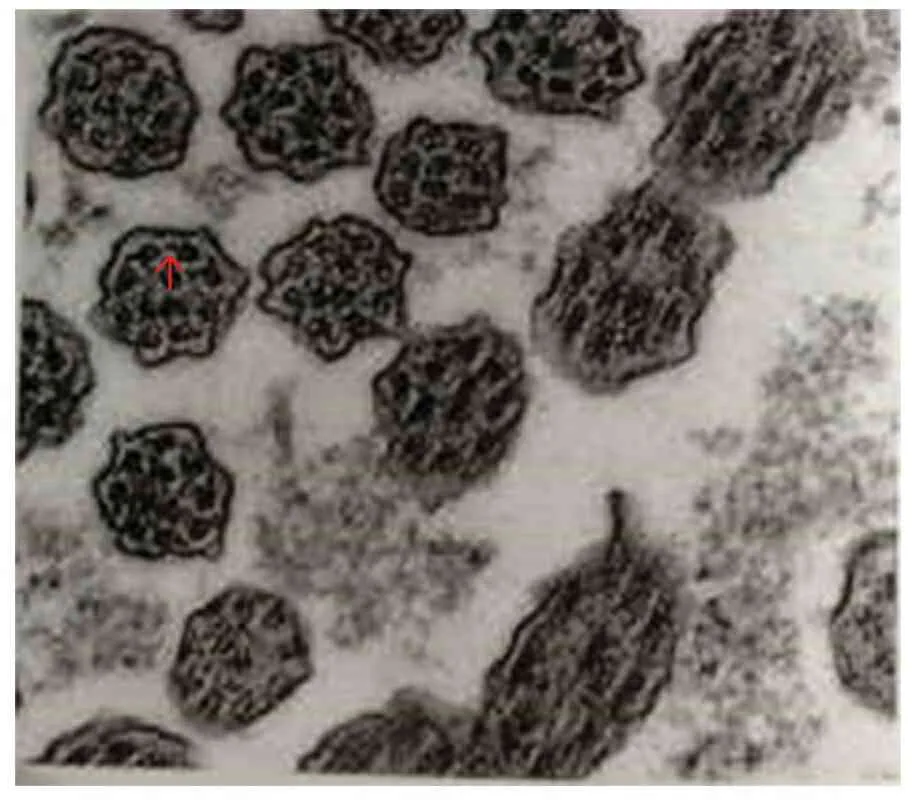
图1 支气管黏膜电镜结果
注 纤毛9+2结构排列紊乱,箭头处示内动力蛋白臂缺失
患儿诊断后予长期治疗及临床随访。接受高渗生理盐水雾化以及拍背物理治疗1年余,2014年后因临床症状好转而自行停用。期间(2013年)因中耳炎行鼓膜置管手术治疗,也因肺炎于当地医院多次住院治疗。2015年起呼吸道感染次数减少,咳嗽、咳痰好转。2016年随访肺功能结果显示,FVC占预计值百分比(60.9 %vs 82.6%)及FEV1占预计值百分比(49.6% vs 77.2%)均较诊断PCD时显著下降;1s率、最大呼气中期流量等也较诊断PCD时明显下降(表1)。胸部CT随访结果显示,与诊断时相比,支气管壁增厚及散在扩张均进展,双肺透亮度不均匀(图2)。
患儿家属有2胎要求,为进一步明确致病基因及进行遗传咨询,在获得患儿家长知情同意下,对患儿及其父母进行了全外显子组测序(WES),并根据基因检测结果进行产前诊断。
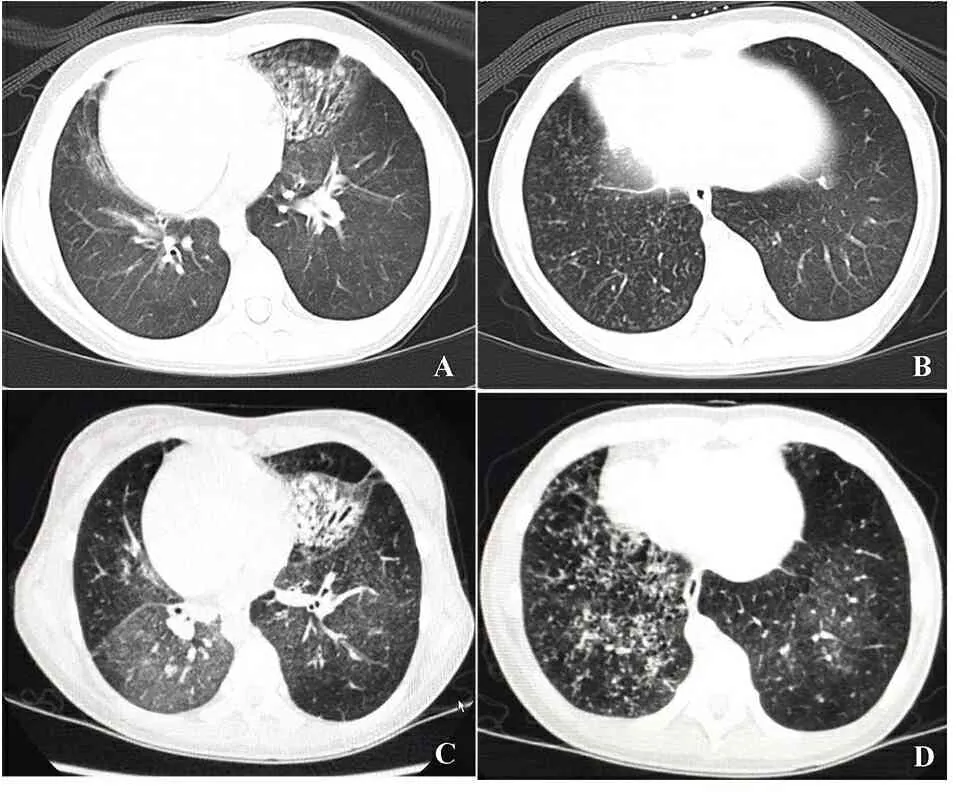
图2 胸部CT随访情况
注 与诊断PCD时(图A和B)比较,2016年7月(图C和D)胸部CT显示肺透亮度不均匀,支气管扩张较前加重
1.2 基因检测及结果 用EDTA抗凝管分别采集患儿及父母静脉全血2 mL,采用mini blood全血试剂盒(德国Qlagen公司)及其标准DNA抽提方法提取基因组DNA,用美国Thermo Fisher公司的NanoDrop紫外分光光度仪测定样本的浓度及定量。参照SureSelect Human All Exon试剂盒(美国Agilent公司)说明书,基因组DNA经过超声打断、末端修复、接头连接、杂交捕获。捕获文库采用Illumina HiSeq 2000平台行WES检测。原始数据经过拼接、比对形成包含变异位点基本信息的vcf文件。按照高通量测序数据分析和临床诊断流程(复旦流程)筛选致病性突变[1]。检测变异采用ANNOVAR和VEP软件参照HG19人类基因组参考序列进行注释,通过千人基因组计划(1000 Genomes)、ExAC及内部数据库注释突变频率,运用SIFT、Polyphen2和MutationTaster在线软件,预测氨基酸替换对蛋白质功能的影响。对检测到的候选突变,采用Primer3在线设计引物,使用Takara公司LA-Taq进行PCR扩增反应,Sanger双向测序(3500 XL Genetic Analyzer, ABI)进行验证。
经WES测序及数据分析,本文病例检测到CCDC39基因c.1819A>A/T, p.K607X无义突变和c.2447_2448het_delCA, p.T816Kfs*3移码突变,查阅文献及OMIM和HGMD等人类疾病突变数据库,发现2个突变均未见文献报道,且2个突变在公共变异频率数据库1 000 genomes和ExAc中均不存在,提示为极其罕见的突变,经功能预测工具MutationTaster均为致病突变,其中CCDC39: c.1819A>A/T, p.K607X无义突变可使翻译提前终止而致蛋白截短,CCDC39: c.2447_2448het_delCA, p.T816Kfs*3移码突变可引起多肽链氨基酸序列的巨大改变。分析家系内突变来源,CCDC39: c.1819A>A/T, p.K607X无义突变来自患儿父亲,CCDC39: c.2447_2448het_delCA, p.T816Kfs*3移码突变来自患儿母亲,属复合杂合突变,符合常染色体隐性遗传模式(图3)。在全基因组基因编码区及周围内含子区未发现其他与患儿临床表型相关的已知或可疑致病基因突变。
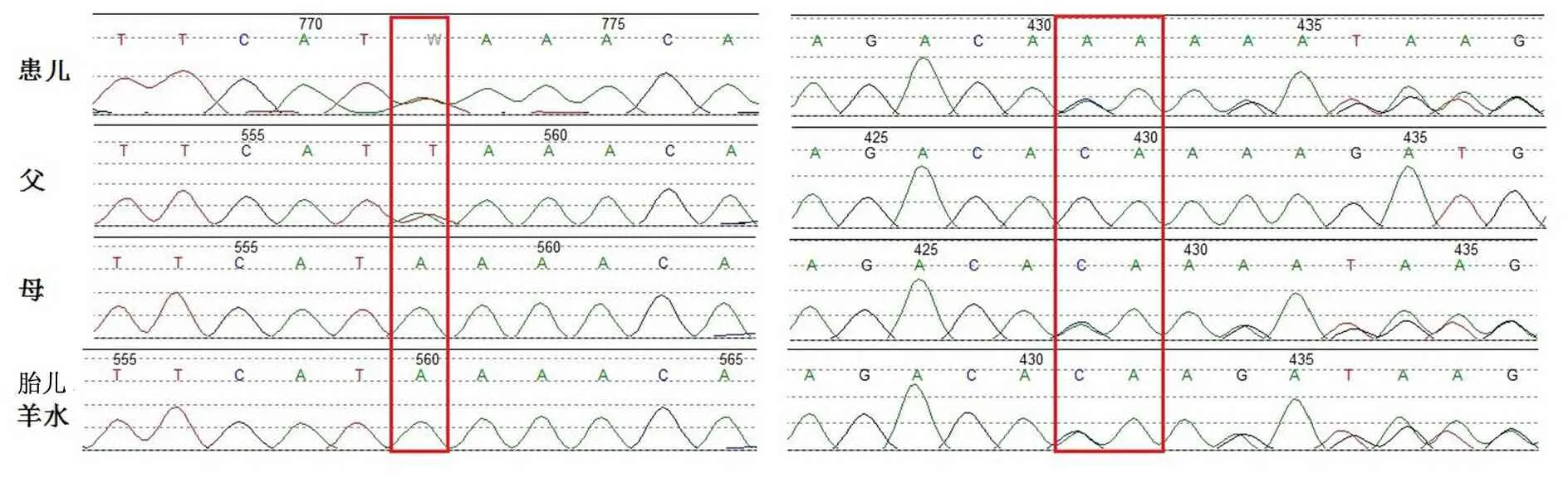
图3 患儿CCDC39基因突变位点Sanger测序验证及家系突变位点验证
注 左图CCDC39: c.1819A>A/T, p.K607X无义突变;右图CCDC39: c.2447_2448het_delCA, p.T816Kfs*3移码突变
1.3 遗传咨询及产前基因诊断 根据患儿检测到的致病基因突变位点的情况向家属介绍CCDC39基因突变所致PCD的临床特征、遗传模式等基因诊断和产前基因诊断的相关问题。从理论预测,母亲再次怀孕胎儿携带复合杂合突变从而致病的概率为1/4,建议行胎儿产前诊断。建议父母的兄弟姐妹等携带致病基因突变的高危人群进行遗传咨询及基因检测。对于携带突变基因者,均应进行下一代基因检测及孕前遗传咨询。告知PCD致病基因众多,有可能其他致病基因自发突变,引起疾病,目前的产前诊断无法预测。经过上述遗传咨询后,患儿母亲要求孕20周前进行该家系的2个突变位点的产前基因诊断。
产前基因诊断胎儿基因突变分析采用羊水细胞基因组DNA。患儿母亲签署知情同意书并获得复旦大学附属儿科医院医学伦理委员会批准,于20 孕周至上海集爱遗传与不孕诊疗中心行超声引导下经腹羊水穿刺,留取羊水10 mL,3 000 rpm离心10min,分离脱落细胞,采用QIAGEN公司DNeasy Blood & Tissue试剂盒提取胎儿基因组DNA。使用LA-Taq酶PCR 扩增CCDC39基因2个突变位点所在的外显子,Sanger双向测序(3500 XL Genetic Analyzer, ABI)。结果显示,胎儿携带母亲来源的CCDC39: c.2447_2448het_delCA, p.T816Kfs*3移码突变,而另一条染色体无父亲的CCDC39: c.1819A>A/T, p.K607X无义突变,为杂合突变携带者,理论预测不会出现PCD的临床表现(图3)。
2 讨论
本文采用WES明确了1例PCD病例的致病基因,发现CCDC39基因复合杂合突变,2个均为未报道的新突变,分别为无义突变和移码突变,可导致编码蛋白截短或氨基酸顺序错乱,对蛋白功能影响巨大。而CCDC39基因突变的病例电镜下可呈现特征性的纤毛微管结构紊乱和内动力蛋白臂缺陷[2,3]。可明确该新突变为致病突变,本文病例CCDC39基因突变致PCD诊断明确。
文献报道的CCDC39基因突变占总PCD病例的4%~9%[4]。Merveille 等[5]研究显示,22例CCDC39基因突变导致的PCD病例中,9例(41%)为内脏正位,10例(45%)为内脏转位,3例(14%)为内脏异位(其中2例为Ivemark综合征)。本文病例表现为伴内脏转位的Kartagener综合征。
PCD是一组遗传异质性疾病,多数认为属常染色体隐性遗传病,也有性染色体遗传的报道。临床表现主要包括新生儿期的呼吸窘迫,儿童及成年期反复呼吸道感染、鼻窦炎、中耳炎、支气管扩张和不孕等。PCD患儿中约50%合并内脏转位,称为Kartagener综合征。PCD常可造成不可逆性肺损伤,可能进展为呼吸衰竭。PCD的诊断主要根据临床表现和诊断性检查,包括影像学、鼻黏膜NO水平、纤毛摆动频率及电镜下观察纤毛的结构异常及功能异常[6]。既往PCD的确诊主要靠电镜下观察到典型的纤毛结构异常[7]。随着遗传学诊断技术的进步,二代测序Panel和WES的应用[8],新的PCD致病基因不断被发现[9]。2016年欧洲呼吸学会最新的PCD的诊断指南中指出了基因诊断在PCD中的重要作用[10]。一方面,部分临床表现典型而电镜下纤毛结构未见异常的病例存在PCD致病基因突变[11],通过分子诊断与纤毛结构分析相结合可以使诊断率从57%增加到69%[12]。另一方面,研究发现电镜下纤毛结构异常与存在缺陷的致病基因密切相关[13,14],而特定的基因类型与PCD肺部病变严重度及预后相关。与本研究结果一致,研究发现与外动力臂缺损及内外动力臂同时缺损相关的基因异常相比,CCDC39及CCDC40基因突变的PCD患儿肺功能更差、胸部CT支气管扩张更明显[15-17]Marthin等[18]的一项74例PCD患儿的横断面纵向研究显示,34%患儿肺功能下降10%以上,57%的患儿肺功能保持相对稳定,还有10%的患儿肺功能有改善,且肺功能与诊断年龄及诊断时的基础水平无明显相关性。一项151例的成人PCD长期随访研究报道肺功能随年龄而呈逐渐下降趋势,FEV1每年下降0.49%,诊断年龄与肺功能下降呈正相关[19]。这些研究结果存在矛盾,可能与研究人群的基因异质性有关。但这些研究均提示基因类型可作为疾病严重度评估和个体化干预治疗的指导指标。
遗传咨询在遗传病的诊疗中起着不可或缺的作用,是理解遗传病发生和预防再发的重要手段,主要内容包括对疾病做出正常的诊断、分析发病原因和遗传方式、家族中携带或再发疾病的风险率、疾病的预后、治疗方法等几个问题。由于PCD无特效治疗,且会累及多个系统,造成呼吸衰竭、不孕和脑积水等严重并发症,国外推荐对PCD患者及亲属进行遗传咨询[4]。因此,对于临床诊断PCD的病例,进行基因诊断来明确致病基因,对于遗传咨询是首要的环节,有重要意义。遗传咨询包括对明确基因诊断先证者的遗传咨询,以及对可能携带者的风险咨询及检测,包括先证者父、母、兄弟、姐妹,先证者父母的兄弟和姐妹等[20]。孕前咨询对发现高遗传疾病风险夫妇,提供孕前筛查等至关重要[21],对降低遗传性疾病的发生起着关键的作用。本研究在先证者临床及基因诊断明确的基础上,对家庭进行了遗传咨询,并对孕妇进行了产前诊断,有益于优生优育。
PCD尚无特异性的治疗手段,现有的治疗方法借鉴于囊性纤维化及支气管扩张的治疗[22,23]。有研究收集DNAH11基因突变的PCD病例的纤毛细胞培养后,采用基因编辑技术对突变病例的纤毛细胞进行位点特异性重组,结果显示可以恢复突变细胞的功能[24]。这项研究为PCD的治疗带来了希望。
本文对PCD的基因诊断、遗传咨询和产前诊断进行了初探,随着对罕见疾病的认识和基因检测、治疗技术的发展,PCD的临床诊疗将会进一步规范,而遗传咨询的规范和普及将对优生优育起到重要作用。
[1]黎籽秀, 刘博, 杨琳, 等. 高通量测序数据分析和临床诊断流程对新生儿多发畸形候选变异的筛选准确性研究. 中国循证儿科杂志. 2015,10(1): 25-28
[2]Blanchon S, Legendre M, Copin B, et al. Delineation of CCDC 39/CCDC40 mutation spectrum and associated phenotypes in primary ciliary dyskinesia. J Med Genet, 2012, 49(6): 410-416
[3]Antony D, Becker-Heck A, Zariwala MA, et al. Mutations in CCDC 39 and CCDC40 are the major cause of primary ciliary dyskinesia with axonemal disorganization and absent inner dynein arms. Hum Mutat, 2013, 34(3): 462-742
[4]Zariwala MA, Knowles MR, Leigh MW. Primary ciliary dyskinesia. //Pagon RA, Adam MP, Bird TD, et al, editors. Gene Reviews. Seattle(WA): University of Washington, Seattle, 1993-2016
[5]Merveille AC, Davis EE, Becker-Heck A, et al. CCDC 39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat Genet, 2011, 43(1): 72-78
[6]Leigh MW, O'Callaghan C, Knowles MR. The challenges of diagnosing primary ciliary dyskinesia. Proc Am Thorac Soc, 2011, 8(5): 434-437
[7]Barbato A, Frischer T, Kuehni CE, et al. Primary ciliary dyskinesia: a consensus statement on diagnostic and treatment approaches in children. Eur Respir J, 2009, 34(6): 1264-1276
[8]Neveling K, Feenstra I, Gilissen C, et al. A post-hoc comparison of the utility of sanger sequencing and exome sequencing for the diagnosis of heterogeneous diseases. Hum Mutat, 2013, 34(12): 1721-1726
[9]Kurkowiak M, Zietkiewicz E, Witt M. Recent advances in primary ciliary dyskinesia genetics. J Med Genet, 2015, 52(1): 1-9
[10]Lucas JS, Barbato A, Collins SA, et al. European Respiratory Society guidelines for the diagnosis of primary ciliary dyskinesia. Eur Respir J, 2016, pii: ERJ-01090-2016
[11]Knowles MR, Leigh MW, Carson JL, et al. Genetic Disorders of Mucociliary Clearance Consortium. Mutations of DNAH11 in patients with primary ciliary dyskinesia with normal ciliary ultrastructure. Thorax, 2012, 67(5): 433-441
[12]Kim RH, A Hall D, Cutz E, et al. The role of molecular genetic analysis in the diagnosis of primary ciliary dyskinesia. Ann Am Thorac Soc, 2014, 11(3): 351-359
[13]Horani A, Brody SL, Ferkol TW. Picking up speed: advances in the genetics of primary ciliary dyskinesia. Pediatr Res, 2014, 75(1-2): 158-164
[14]Knowles MR, Daniels LA, Davis SD, et al. Primary ciliary dyskinesia. Recent advances in diagnostics, genetics, and characterization of clinical disease. Am J Respir Crit Care Med, 2013, 188(8): 913-922
[15]Davis SD, Ferkol TW, Rosenfeld M, et al. Clinical features of childhood primary ciliary dyskinesia by genotype and ultrastructural phenotype. Am J Respir Crit Care Med, 2015, 191(3): 316-324
[16]Wallmeier J, Al-Mutairi DA, Chen CT, et al. Mutations in CCNO result in congenital mucociliary clearance disorder with reduced generation of multiple motile cilia. Nat Genet, 2014, 46(6): 646-651
[17]Boon M, Wallmeier J, Ma L, et al. MCIDAS mutations result in a mucociliary clearance disorder with reduced generation of multiple motile cilia. Nat Commun, 2014, 5: 4418
[18]Marthin JK, Petersen N, Skovgaard LT, et al. Lung function in patients with primary ciliary dyskinesia: a cross-sectional and 3-decade longitudinal study. Am J Respir Crit Care Med, 2010, 181(11): 1262-1268
[19]Shah A, Shoemark A, MacNeill SJ, et al. A longitudinal study characterising a large adult primary ciliary dyskinesia population. Eur Respir J, 2016, 48(2): 441-450
[20]Lucas JS, Paff T, Goggin P, et al. Diagnostic Methods in Primary Ciliary Dyskinesia. Paediatr Respir Rev, 2016, 18: 8-17
[21]Metcalfe SA. Carrier screening in preconception consultation in primary care. J Community Genet, 2012, 3(3): 193-203
[22]Hosie P, Fitzgerald DA, Jaffe A, et al. Primary ciliary dyskinesia: overlooked and undertreated in children. J Paediatr Child Health, 2014, 50(12): 952-958
[23]Lucas JS, Carroll M. Primary ciliary dyskinesia and cystic fibrosis: different diseases require different treatment. Chest, 2014, 145(4): 674-676
[24]Lai M, Pifferi M, Bush A, et al. Gene editing of DNAH11 restores normal cilia motility in primary ciliary dyskinesia. J Med Genet, 2016, 53(4): 242-249
(本文编辑:张崇凡,孙晋枫)
GeneticcounselingandprenataldiagnosisforprimaryciliarydyskinesiainacasecausedbyCCDC39genemutations
QIYuan-yuan1,HONGDa1,WANGHui-jun,ZHANGXiao-bo,WANGLi-bo,JINTing-ting,QIANLi-ling
(Children'sHospitalofFudanUniversity,Shanghai201102,China)
Corresponding Author:QIAN Li-ling,E-mail: llqian@126.com; JIN Ting-ting
ObjectiveTo identify the genetic defect in a child with primary ciliary dyskinesia and provide genetic counseling and prenatal diagnosis for the family. MethodsWhole exome sequencing (WES) was performed in a case with a clinical diagnosis of Kartagener syndrome as well as her asymptomatic parents. The data analysis pipeline of Children's Hospital of Fudan University was used to identify pathogenic mutations. Amniocentesis was conducted for the mother in her second pregnancy after a thorough genetic counseling. Genomic DNA was extracted from exfoliated amniotic fluid cells and Sanger sequencing was performed after PCR amplification. ResultsDuring follow-ups, the lung function of the patient deteriorated markedly and bronchiectasis shown in chest CT was aggravated. A heterozygous nonsense mutation (c.1819A>A/T, p.K607X) and a frameshift mutation (c.2447_2448het_delCA, p.T816Kfs*3) inCCDC39 gene were detected in this patient by WES. Both mutations were novel and predicted to be disease-causing. Mutational analysis of the parents demonstrated they were compound heterozygous mutations. These compound heterozygous mutations were consistent with ciliary structural abnormity revealed by electron microscope thus suggesting the pathogenic nature of these mutations. Sequencing of the amniotic fluid cells showed that the fetus only carried one heterozygous mutation which was inherited from the mother.ConclusionThe compound heterozygous mutations inCCDC39 gene detected by WES was the genetic cause of primary ciliary dyskinesia. Genetic counseling and prenatal diagnosis may be helpful to the family based on the basis of this genetic diagnosis.
Primary ciliary dyskinesia; Whole exome sequencing ; Genetic counseling; Prenatal diagnosis
上海市科委西医引导项目:134119a7800、14411962102;上海市人才发展基金:沪人201450
复旦大学附属儿科医院 上海,201102;1 共同第一作者
钱莉玲,E-mail: llqian@126.com;金婷婷,E-mail:super_jint t@163.com
10.3969/j.issn.1673-5501.2016.06.009
2016-08-22
2016-12-13)
猜你喜欢
自然杂志(2022年3期)2022-08-18
医学研究生学报(2021年4期)2021-12-02
临床与实验病理学杂志(2021年3期)2021-04-25
中国生殖健康(2020年2期)2021-01-18
中华皮肤科杂志(2019年5期)2019-06-24
小学生导刊(2018年13期)2018-06-29
中国生殖健康(2018年2期)2018-01-12
中国工程咨询(2017年3期)2017-01-31
中国神经精神疾病杂志(2014年1期)2014-03-01
祝您健康(1987年2期)1987-12-30
