
Electronic Structures and Thermoelectric Properties of Two-Dimensional MoS2/MoSe2Heterostructures
2016-09-23 06:06Tian-minWu,Rui-xueXu,XiaoZheng等
Electronic Structures and Thermoelectric Properties of Two-Dimensional MoS2/MoSe2Heterostructures
I.INTRODUCTION
Thermoelectric materials are considered to have great potential for power generation,energy saving,and environmental protection[1-5].Various thermoelectric semiconductor materials,such as chalcogenides[6,7],zintl phases[8],clatharates[9],complex oxides[10],and skutterudites[11,12]have been developed to convert the waste heat into electricity.The efficiency of thermoelectric device is measured by the material-dependent figure of merit(ZT=S2σT/κ)[13].Here,S is the Seebeck coefficient,σ is the electrical conductivity,and κ=κe+κlatis the thermal conductivity(κeand κlatrepresent the electronic and lattice thermal conductivity contributions,respectively).Ⅰn general,the figure of merit(ZT)can be enhanced by improving the power factor(S2σ)or decreasing the thermal conductivity(κ) of material.The search of materials of high thermoelectric performance is confronted with the challenge that electronic conductivity and Seebeck coefficient have opposite dependencies on the materials parameters:electronic conductivity(σ)increases as the doping level is improved,but higher doping level leads to lower Seebeck coefficient.Various theoretical and experimental methods have been introduced to enhance the value of ZT[14-26].For instance,rattler atoms implanted into open structures like clatharates[27]and skutturides[28] have been introduced to have low-frequency phonons near the acoustic branches,thus enhancing the phonon scattering to reduce the lattice thermal conductivity of the materials.
Two-dimensional(2D)transition-metaldichalcogenide semiconductors such as MoS2and MoSe2have significant potential as the ideal thermoelectric materials,since they possess low thermal conductivity along the direction perpendicular to the lattice layers[29-33]. Despite this advantage,ZT values of transition-metal dichalcogenides(TMDCs)are still low due to the difficulty of enhancing the power factor[34-37].To solve this problem,we propose the use of 2D heterostructures systems such as MoS2/MoSe2,which are formed by stacking two different TMDCs layers together.Based on density functional theory(DFT)calculations,we demonstrate that such 2D heterostructures materials possess many improved thermoelectric properties as compared with their parental pristine materials.
II.COMPUTATIONAL METHODS
First-principlescalculationsofbulkandbilayer MoS2/MoSe22D heterostructures system are conducted using DFT methods implemented in the Vienna ab initio simulation package(VASP)[38].The projector-augmented-wave pseudopotentials and the generalized gradient approximation of Perdew,Burke,and Ernzerhof(PBE)for exchange-correlation functional are adopted in our simulations[39].Furthermore,van der Waals interactions are taken into account by us-ing the semiempirical correction of Grimme(DFT-D3) [40].The energy cutoff for a plane-wave expansion is set to 500 eV.All atomic coordinates are relaxed until the atomic forces have declined to 0.01 eV/˚A,enforcing a total energy convergence criterion of 1×10-5eV.A vacuum slab larger than 15˚A is added to avoid interaction between adjacent images of the bilayer structure.
Temperature-and doping-dependent electronic transport properties,including electronic conductivity and Seebeck coefficient,are computed by using the semiclassical Boltzmann transport theory[41,42].The constant scattering time approximation is adopted,which is valid if the electron relaxation time does not very strongly with the energy on a scale of kBT,and the rigid band approaches as implemented in the BoltzTraP code [43].By using a Fourier expansion[44,45],while maintaining the crystal symmetry[46],the BoltzTrap code fits the ab initio electronic band structure to an analytic function.Since the transport properties can be very sensitive to the Brillouin zone(BZ)sampling,especially for low doping levels and low temperatures,we calculate the electronic structures required for the transport calculations with very dense k-meshes(43×43×11 for the hexagonal BZ of bulk 2D heterostructures,and 43×43×5 for the trigonal BZ of the bilayer structure).
Thetemperature-dependentanddoping-leveldependent thermoelectric transport tensors,such as electronic conductivity σαβ(T,µ),Seebeck coefficient Sαβ(T,µ),and thermal conductivity(electronic part κel
αβ)tensors are calculated as follows[43]:
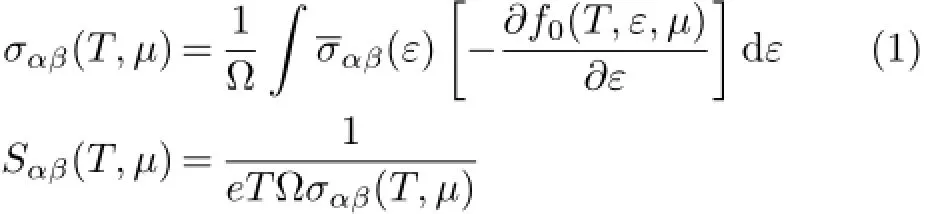
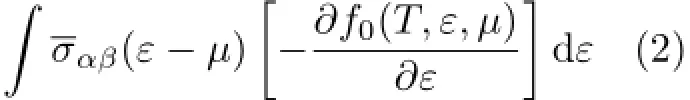
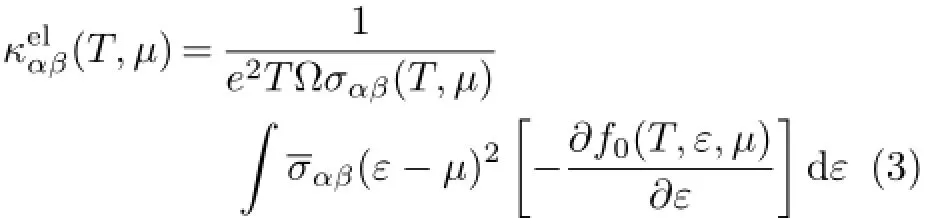
Here,α and β are the tensor indices,Ω is the volume of the unit cell,e is the charge of the electron,f0(T,ε,µ) is the Fermi distribution function,andµis the Fermi level.The conductivity tensor σαβ(ε)(transport distributions)can be expressed analytically as[43]:
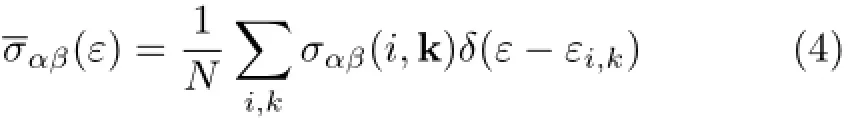
which can be expressed using k-depedent conductivity tensor as[43]:
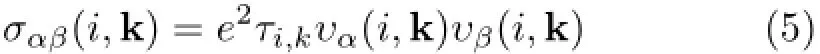
where i is the band index,k is the reciprocal vector,N is a normalization depending on the number of k points sampled in the BZ,τi,kis the electronic relaxation time,and υα(i,k)is the i component of band velocity▽kε(k)with ε being the band energies.As demonstrated in the above formula,temperature-dependent transport properties can be calculated from the Fermi distribution function,and one can achieve the dopinglevel dependent transport properties by adjusting the Fermi levelµwhich is directly associated with doping concentration[43].
The electron relaxation time τ characterizing the average time between two consecutive electron scattering events is a crucial parameter for calculating the thermoelectric properties,such as the electrical conductivity(σ)and the electronic thermal conductivity(κe),since just the ratio of these conductivity to the relaxation time(σ/τ and κe/τ)can be achieved by using the BoltzTrap code.The value of τ is usually obtained by fitting the calculated ratio of σ/τ to measure electrical conductivity data.However,such experimental data for the MoS2/MoSe22D heterostructures have so far remained unavailable.Therefore,in this work we focus on the ratio of thermal properties to the electron relaxation time rather than on the properties themselves.
III.RESULTS AND DISCUSSION
A.Crystal structure
The monolayer structure of MoS2and MoSe2,consists of a single(S/Se)-Mo-(S/Se)layer with space group P6m2(187),which has no inversion symmetry. While the bulk MoS2and MoSe2has a 2H-polytype structure in the P63/mmc space group(194).Ⅰts supercell consists of two(S/Se)-Mo-(S/Se)layers separated along the z axis,and the two layers are bound by van der Waals interactions.Furthermore,due to the increasing radius of the chalcogen atoms,the optimized values of lattice constants for mo˚nolayer alongin-plan˚e direction increase from a=3.162A in MoS2to3.320A in MoSe2.Ⅰn general the lattice mismatch may lead to stacking disorder or Moir´e Pattern superstructures.However,the intrinsic lattice mismatch between the layer of MoS2and MoSe2is as small as 0.158˚A. Moreover,the explicit consideration of such a small lattice mismatch would require the use of very large supercell and thus make the calculation rather expensive. Therefore,in our simulations for the 2D heterostructures,the same lattice constant is adopted for the both types of layers(MoS2and MoSe2).
The optimized MoS2/MoSe22D heterostructures for bulk system in our calculation has the lattice parameter of a=3.248 and 3.250˚A for bilayer,which is consistent with the other theoretical results[47-49].As in Fig.1,the Mo atoms of MoSe2monolayer sit on the top of the chalcogen atoms S of MoS2monolayer,and those monolayer which constructs the heterostructures shows a lateral shift.Due to the van der Waals binding,the Moatoms between different layers are separated by 6.36˚A in both bulk and bilayer systems.To further study the electronic properties and thermoelectric properties of bulk and bilayer MoS2/MoSe2heterostructures,their band structures and density of states(DOS)are calculated.woqujieninmen
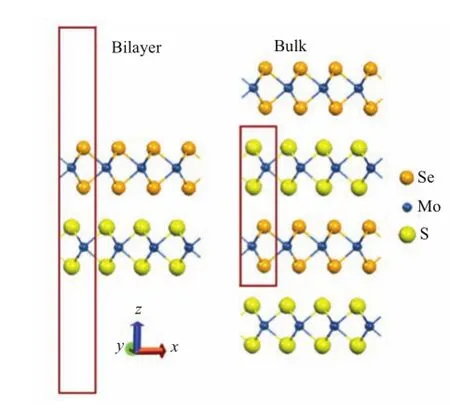
FⅠG.1 Atomic structure of bulk and bilayer MoS2/MoSe22D heterostructures.The Se,Mo,and S atoms are represented by orange,blue,and yellow colors,respectively.The red box represents the unit cell used in our simulations.
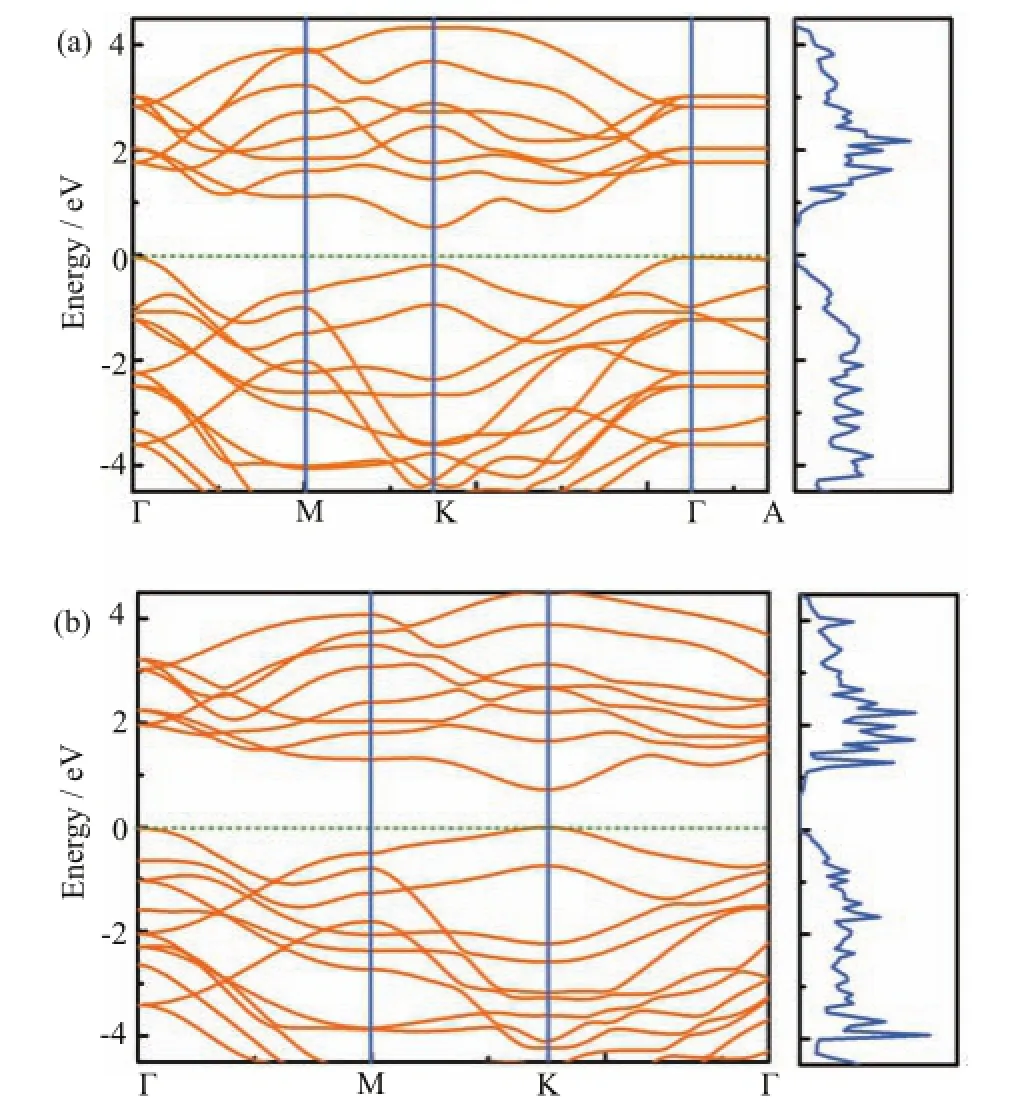
FⅠG.2 Electronic band structures(left)and DOS(right) calculated by using PBE along the high-symmetry lines for (a)bulk and(b)bilayer MoS2/MoSe2heterostructures.
B.Electronic structure
Electronic band structures and DOS for bulk and bilayer MoS2/MoSe22D heterostructures are presented in Fig.2.For the bulk 2D heterostructures,as demonstrated in Fig.2(a),an indirect band gap of 0.546 eV is observed with the valence band maximum at the Γ point and the conduction band minimum at the K point,which is consistent with the theoretical results reported by Changhoon et al.[47].The electronic band structure along high symmetry point of in-plane and crossplane direction shows a great anisotropy due to the 2D heterostructures constructed by van der Waals binding,and the band gap of the in-plane bands(Γ-M-K-Γ)is much smaller than cross-plane one(Γ-A).Bilayer 2D heterostructures,on the other hand,also show an indirect band gap of 0.695 eV with both the valance band maximum at the Γ point and conduction band minimum at the high symmetry K point,which is consistent with other theoretical results[49].Furthermore,both the bulk and bilayer systems show a strong asymmetric feature between the valence and conduction bands. Although GGA-PBE is known to underestimate band gaps of semiconductors,the resulting electronic structure are considered to be reasonably accurate for subsequent computation of thermoelectric properties.
C.Thermoelectric properties
To enlarge ZT,the material should have a larger power factor S2σ and smaller κ(κ=κe+κlat).Figure 3 depict the in-plane and cross-plane power factor divided by the relaxation time τ,S2σ/τ,of the bulk MoS2/MoSe2heterostructures under doping(both pand n-type),with the temperature ranging from 300 K to 1200 K.Here,τ is the relaxation time that is not directly determined by the band structure,but depends on the temperature,the doping level,and also the sample details(such as defect types and concentrations) [43].Note that we compare S2σ/τ instead of S2σ,because the relaxation time is difficult to calculate and the electronic conductivity has not been measured experimentally.As demonstrated in Fig.3,S2σ/τ is enhanced as the carrier concentration increases at each temperature.Ⅰts maximum value is nearly within the carrier concentration range of 1020-1021cm-3for each temperature,which also implies that high carrier concentration could enhance the power factor.As shown in Fig.3,for bulk heterostructures,the p-type doping at in-plane direction shows the largest power factor S2σ/τ at temperature 1200 K.To better reflect the thermoelectric performance of bulk 2D MoS2/MoSe2heterostructures,the theoretical results of pristine bulk MoSe2at the hole carrier concentration of 5×1020cm-3are adopted [36].As demonstrated in TableⅠ,although the pristine MoSe2shows a slightly higher S2σ/τ at 1200 K,the bulk MoS2/MoSe2heterostructures shows an overall better thermoelectric performance along the in-plane direction.Despite the difference in relaxation time between these two crystals,such a comparison provides an overall assessment for their thermoelectric performances.
Along the cross-plane direction,the n-type doping shows a greater power factor S2σ/τ than p-type one. However,since in TMDCs the relaxation time of inplane direction is two orders of magnitude larger than the cross-plane one[36,37],the in-plane electrical conductivity is typically two orders magnitude larger than the cross-plane electrical conductivity.Then,the power factor along in-plane direction is expected to be two orders of magnitude larger than the cross-plane counterpart,since both of them have similar Seebeck coefficient values.Additionally,the DOS close to the valence band edge is much larger than those near the conduction band edge,which suggests that p-type doping could have a better thermoelectric performance[50].Therefore,we will focus on the p-type doping bulk heterostructures,and discuss the temperature-dependent and dopinglevel-dependent Seebeck coefficient,the electric conductivity and the thermal conductivity individually.
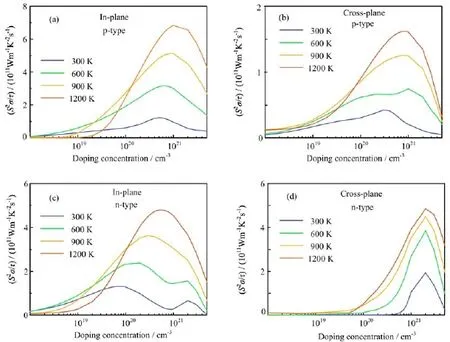
FⅠG.3Ⅰn-plane(a,c)and cross-plane(b,d)temperature-dependent power factor divided by the scattering time(PF/τ)as a function of p-and n-type doping concentration for bulk MoS2/MoSe22D heterostructures.
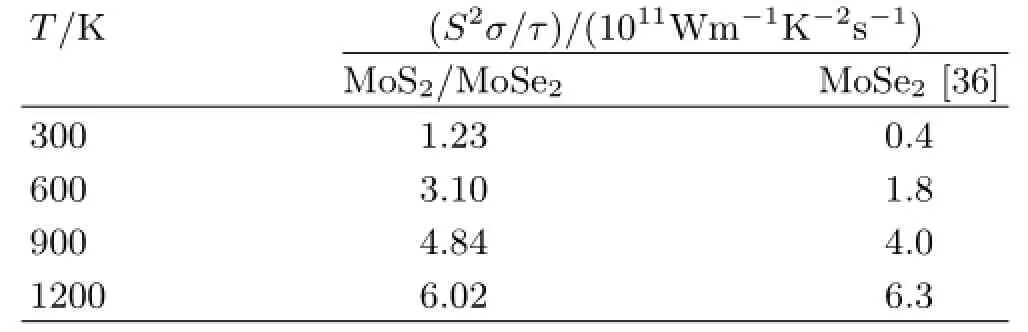
TABLEⅠTemperaturedependentpowerfactordivided with relaxation time(S2σ/τ)for bulk MoSe2and MoS2/MoSe22D heterostructures along the in-plane direction at the hole carrier concentration of 5×1020cm-3.
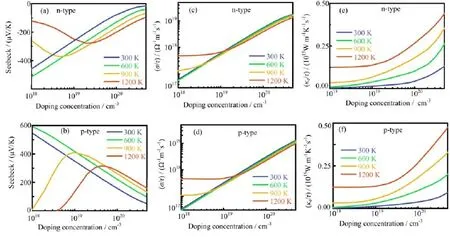
The p-type doping in-plane and cross-plane Seebeck coefficients as functions of carrier concentration are shown in Fig.4(a)and(b).Similar to the other TMDCs [36,37],the bulk 2D heterostructures has a large Seebeck coefficient.At a fixed carrier concentration,for both in-plane and cross-plane directions,the Seebeck coefficient of bulk 2D heterostructures slightly increases as the temperature is increasing.This phenomenon mainly originates from the Fermi broadening as the temperature rises,and then it leads to an increasing effective density of states at the top of valence band [51].Consistent with the known thermoelectric behavior for the other TMDCs[36,37],the maximum value of Seebeck coefficient for each temperature shifts to high doping level and decrease as temperature is increased for both cross-and in-plane direction.The calculated tiny band gap for bulk 2D heterostructures(0.546 eV),as mentioned above,likely lead to the bipolar effect at low doping carrier concentration which make the Seebeck coefficient decreases with decreasing doping concentration,opposite to the usual situation[52].As temperature is higher than 900 K,the Seebeck coefficient along the in-plane direction presents a sign reversal at low doping level,which is attributed to the increasing negative contribution of thermally excited electrons to the Seebeck coefficient under the bipolar-transport conditions[53].While the Seebeck coefficient along the cross-plane direction does not show a sign reversal and more stable with the doping concentration increasing at low doping level,indicating that the system along cross-plane direction shows a weak bipolar effect originated by the large band gap along this direction.This anisotropy can also be observed by the electrical conductivity divided by the relaxation time(σ/τ)as a function of doping concentration.As demonstrated in Fig.4 (c)and(d),the conductivity divided by the relaxation time(σ/τ)of in-plane is about one order higher than the cross-plane one,revealing that the thermally excited carrier concentration along the in-plane direction is significantly higher than cross-plane one.Furthermore,in the 900-1200 K temperature range,the electrical conductivity divided by the relaxation time(σ/τ) does not depend strongly on the doping concentration at low doping level,since the thermally excited carriers dominate transport[53].To overcome the contribution by the thermally excited carrier concentration,the higher doping concentration is required as temperature is higher.As the doping concentration above the onset of bipolar transport,the electrical conductivity divided by the relaxation time(σ/τ)increases substantially with the increasing doping concentration.
The electronic band structure also supports this viewpoint,as the band gap of in-plane(along high symmetry point Γ-M-K-Γ)is much smaller than the crossplane one(Γ-A).The electrical thermal conductivities divided by the relaxation time(κe/τ)along the in-plane and cross-plane directions are depicted in Fig.4(e)and (f),respectively.Although the electronic contribution to the thermal conductivity is small,compared with the lattice thermal conductivity,the electronic thermal conductivity also illustrates the anisotropy of thermal conductivity between cross-plane and in-plane.The power factor of in-plane is considerably larger than the crossplane one,especially since the relaxation time of crossplane is much smaller than the in-plane one.Furthermore,the electronic thermal conductivity divided by the relaxation time(κe/τ)at a fixed carrier concentrations for both in-plane and cross-plane increases as temperature rising,mainly because of more intense electron scattering as temperature increasing.According to the Wiedemann-Franz law[43],

FⅠG.4Ⅰn-plane(a,c,e)and cross-plane(b,d,f)temperature-and p-type doping concentration-dependent Seebeck coefficient,electronic conductivity divided by the scattering time(σ/τ),and electronic thermal conductivity divided by the scattering time(κe/τ)of bulk MoS2/MoSe2heterostructures,respectively.
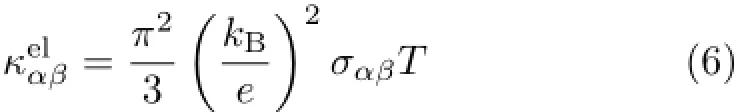
the electronic thermal conductivity is proportional to the temperature of system.Meanwhile,since thermal conductivity is proportional to the electronic conductivity that is highly anisotropic,there is a significantdifference between the in-plane and cross-plane components.
Compared with the bulk heterostructures,the bilayer heterostructures show a much lower power factor (S2σ/τ)as demonstrated in Fig.5.Since the band gap of bilayer 2D heterostructures is larger than the bulk one,the electronic conductivity of bilayer heterostructures is much smaller than that of the bulk,see Fig.6 (a)and(b).Furthermore,since the bulk and bilayer heterostructures have similar Seebeck coefficients,as demonstrated in Fig.6(c)and(d),the bulk heterostructures should have a better thermoelectric performance than those of bilayer one.While the electrical thermal conductivity is smaller than the bulk one,as presented in Fig.6(e)and(f).For p-type doping,the bilayer heterostructures show a similar thermoelectric performance to the monolayer one[36].

FⅠG.5 Temperature-dependent power factor divided by the scattering time(PF/τ)of bilayer MoS2/MoSe22D heterostructures as function of n-and p-type doping concentration.
FⅠG.6 Temperature dependent Seebeck coefficient(a,b),electronic conductivity divided by the scattering time σ/τ(c,d),and electronic thermal conductivity divided by the scattering time κe/τ(e,f)of bilayer MoS2/MoSe2heterostructures as a function of n-and p-type doping concentration,respectively.
IV.CONCLUSION
Based on the electrical band structure calculated from first principles,the thermoelectric properties of bulk and bilayer 2D MoS2/MoSe2heterostructures have been analyzed by using the semi-classical Boltzmann transport theory.Both n-type and p-type doping have been addressed for bulk and bilayer heterostructures,employing the rigid band approximation and constant scattering time approximation.Due to a smaller band gap and more dense DOS close to the valence band edgethan those near the conduction band edge,the thermoelectric performance of bulk 2D MoS2/MoSe2heterostructures turns out to be superior to pristine bulk MoSe2along the in-plane direction for p-type doping at a wide temperature range.Furthermore,with a larger band gap,the bilayer heterostructures show a much lower electronic conductivity than those of bulk heterostructures,which also induces that it shows a weaker thermoelectric performance than bulk one.Although the power factor of bilayer heterostructures is lower than those of bulk heterostructures,it shows a similar thermoelectric performance to the monolayer MoSe2for p-type doping at each temperature.Therefore,we safely conclude that such 2D heterostructures materials possess much improved thermoelectric properties as compared with their parental pristine materials,especially for bulk one.As reported by Li et al.[54],the thermal lattice conductivity considered the spin-orbit coupling(SOC)effect is higher than those value not considered.Furthermore,the band shape and the band gap which make a great influence on the calculation of thermoelectric properties could be changed as the SOC effect is introduced,thus the SOC effect should be taken into account.However,the SOC is not considered in the present work,because of the limited computational resources at our disposal.The influence of the SOC on the thermoelectric properties is to be addressed in our future work.
V.ACKNOWLEDGMENTS
This work was supported by the National NaturalScienceFoundationofChina(No.21203178,No.21373201,No.21433014,No.21233007,No.21303175,and No.21322305),the Science and Technological Ministry of China(No.2011YQ09000505),the“Strategic Priority Research Program”of the Chinese Academy of Sciences(No.XDB10040304 and No.XDB100202002),and the Fundamental Research Funds for the Central Universities(No.2340000074).The computational resources are provided by the Supercomputing Center of University of Science and Technology of China.
[1]M.S.Dresselhaus,G.Chen,M.Y.Tang,R.G.Yang,H.Lee,D.Z.Wang,Z.F.Ren,J.P.Fleurial,and P. Gogna,Adv.Mater.19,1043(2007).
[2]K.Biswas,J.He,Ⅰ.D.Blum,C.Wu,T.Hogan,D.N. Seidman,V.P.Dravid,and M.G.Kanatzidis,Nature 489,414(2012).
[3]K.P.Pernstich,B.R¨ossner,and B.Batlogg,Nat. Mater.7,321(2008).
[4]W.J.Liang,A.Ⅰ.Hochbaum,M.Fardy,O.Rabin,M. J.Zhang,and P.D.Yang,Nano Lett.9,1689(2009).
[5]G.J.Snyder and E.S.Toberer,Nat.Mater.7,105 (2008).
[6]C.Wood,Rep.Prog.Phys.51,459(1988).
[7]L.E.Shelimova,O.G.Karpinskii,P.P.Konstantinov,E.S.Avilov,M.A.Kretova,and V.S.Zemskov,Ⅰnorg. Mater.40,451(2004).
[8]S.M.Kauzlarich,S.R.Brown,and G.J.Snyder,Dalton Trans.2099(2007).
[9]G.S.Nolas,G.A.Slack,and S.B.Schujman,In Recent Trends in Thermoelectric Materials Research I,T.M. Tritt,Ed.,Semiconductors and Semimetals,San Diego,CA:Academic Press,255(2001).
[10]K.Koumoto,Ⅰ.Terasaki,and R.Funahashi,MRS Bull. 31,206(2006).
[11]G.S.Nolas,D.T.Morelli,and T.M.Tritt,Annu.Rev. Mater.Sci.29,89(1999).
[12]C.Uher,In Recent Trends in Thermoelectric Materials Research I,T.M.Tritt,Ed.,Semiconductors and Semimetals,San Diego,CA:Academic Press,139 (2001).
[14]G.A.Slack and D.M.Rowe,CRC Handbook of Thermoelectrics.Boca Raton:CRC Press,40(1995).
[15]L.D.Hicks and M.S.Dresselhaus,Phys.Rev.B 47,12727(1993).
[16]L.D.Hicks and M.S.Dresselhaus,Phys.Rev.B 47,16631(1993).
[17]Y.Wu,R.Fan,and P.Yang,Nano Lett.2,83(2002).
[18]R.Yang,G.Chen,and M.S.Dresselhaus,Phys.Rev. B 72,125418(2005).
[19]C.J.Vineis,A.Shakouri,A.Majumdar,and M.G. Kanatzidis,Adv.Mater.22,3970(2010).
[20]A.Ⅰ.Boukai,Y.Bunimovich,J.Tahir-Kheli,J.K.Yu,W.A.GoddardⅠⅠⅠ,and J.R.Heath,Nature 451,168 (2008).
[21]X.Tang,W.Xie,H.Li,W.Zhao,Q.Zhang,and M. Niino,Appl.Phys.Lett.90,012102(2007).
春节期间,该站点的VOCs关键活性物种主要为丙烯、乙烯、间/对二甲苯、甲苯、正丁烷、异戊烷、异丁烷、反-2-丁烯、丙烷和1-丁烯。
[22]B.Poudel,Q.Hao,Y.Ma,Y.Lan,A.Minnich,B.Yu,X.Yan,D.Wang,A.Muto,D.Vashaee,X.Chen,J. Liu,M.S.Dresselhaus,G.Chen,and Z.Ren,Science 320,634(2008).
[23]T.Ⅰkeda,L.A.Collins,V.A.Ravi,F.S.Gascoin,S.M. Haile,and G.J.Snyder,Chem.Mater.19,763(2007).
[24]R.Venkatasubramanian,E.Siivola,T.Colpitts,and B. O’Quinn,Nature 413,597(2001).
[25]T.Ⅰkeda,S.M.Haile,V.A.Ravi.,H.Azizgolshani,F.Gascoin,and G.J.Snyder,Acta Mater.55,1227 (2007).
[26]T.C.Harman,P.J.Taylor,M.P.Walsh,and B.E. LaForge,Science 297,2229(2002).
[27]J.Dong,O.F.Sankey,and C.W.Myles,Phys.Rev. Lett.86,2361(2001).
[28]V.Keppens,D.Mandrus,B.C.Sales,B.C.Chakoumakos,P.Dai,R.Coldea,M.B.Maple,D.A.Gajewski,E.J.Freeman,and S.Bennington,Nature 395,876 (1998).
[29]L.H.Brixner,J.Ⅰnorg.Nucl.Chem.24,257(1962).
[30]S.H.El-Mahalawy and B.L.Evans,Phys.Status Solidi B 79,713(1977).
[31]E.Revolinsky and D.Beerntsen,J.Appl.Phys.35,2086(1964).
[32]C.Muratore,V.Varshney,J.J.Gengler,J.J.Hu,J.E.Bultman,T.M.Smith,P.J.Shamberger,B.Qiu,X.Ruan,A.K.Roy,and A.A.Voevodin,Appl.Phys. Lett.102,081604(2013).
[33]A.Mavrokefalos,N.T.Nguyen,M.T.Pettes,D.C. Johnson,and L.Shi,Appl.Phys.Lett.91,171912 (2007).
[34]M.Kayyalha,L.Shi,andY.P.Chen,arXiv: 1505.05891(2015).
[35]H.Kedar,W.Ying,Y.Yu,Z.Hanyu,W.Yuan,M. Joel,and Z.Xiang,arXiv:1505.06779(2015).
[36]S.Kumar and U.Schwingenschl¨ogl,Chem.Mater.27,1278(2015).
[37]A.N.Gandi and U.Schwingenschl¨ogl,Chem.Mater. 26,6628(2014).
[38]G.Kresse and J.Furthm¨uller,Phys.Rev.B 54,11169 (1996).
[39]J.P.Perdew,K.Burke,and M.Ernzerhof,Phys.Rev. Lett.77,3865(1996).
[40]S.Grimme and G.G.Semiempirical,J.Comput.Chem. 27,1787(2006).
[41]J.M.Ziman,Principles of the Theory of Solids,Cambridge:Cambridge University Press(1972).
[42]P.B.Allen,W.E.Pickett,and H.Krakauer,Phys.Rev. B 37,7482(1988).
[43]G.Madsen and D.Singh,Comput.Phys.Commun. 175,67(2006).
[44]R.N.Euwema,D.J.Stukel,T.C.Collins,J.S.Dewitt,and D.G.Shankland,Phys.Rev.178,1419(1969).
[45]W.Jones and N.March,Theoretical Solid State Physics:Perfect Lattices in Equilibrium,Dover Books on Physics,New York:John Wiley&Sons,Ltd.,(1973).
[46]D.D.Koelling and J.H.Wood,J.Comput.Phys.67,253(1986).
[47]L.Changhoon,H.Jisook,W.Myung-Hwan,and H.S. Ji,Chem.Mater.25,3745(2013).
[48]T.Humberto,L.Florentino,and T.Mauricio,Sci.Rep. 3,1549(2013).
[49]N.Lu,H.Guo,L.Lei,J.Dai,L.Wang,W.N.Mei,X. Wu,and X.C.Zeng,Nanoscale 6,2879(2014).
[50]Z.Lijun and J.S.David,Phys.Rev.B 81,245119 (2010).
[51]Z.Gang and W.Dong,Sci.Rep.5,8099(2015).
[52]S.Bao-Zhen,M.Zuju,H.Chao,and W.Kechen,RSC Adv.5,56382(2015).
[53]G.Shi and E.Kioupakis,J.Appl.Phys.117,065103 (2015).
[54]R.Li,X.Cheng,Q.Xie,Y.Sun,D.Li,Y.Li,and X. Chen,Sci.Rep.5,8466(2015).
Tian-min Wua,Rui-xue Xua,Xiao Zhenga∗,Wei Zhuangb∗
a.Hefei National Laboratory for Physical Sciences at the Microscale&Synergetic Innovation Center of Quantum Information and Quantum Physics,University of Science and Technology of China,Hefei 230026,China
b.State Key Laboratory of Structural Chemistry,Fujian Institute of Research on the Structure of Matter,Chinese Academy of Sciences,Fuzhou 350002,China
(Dated:Received on December 30,2015;Accepted on March 15,2016)
Thermoelectric properties of bulk and bilayer two-dimensional(2D)MoS2/MoSe2heterostructures are investigated using density functional theory in conjunction with semiclassical Boltzmann transport theory.Ⅰt is predicted that the bulk 2D heterostructures could considerably enhance the thermoelectric properties as compared with the bulk MoSe2.The enhancement originates from the reduction in the band gap and the presence of interlayer van der Waals interactions.We therefore propose the 2D MoS2/MoSe2heterostructures as a possible candidate material for thermoelectric applications.
Key words:Heterostructures,Thermoelectric property,Density functional theory,Boltzmann transport theory
∗Authors to whom correspondence should be addressed.E-mail: xz58@ustc.edu.cn,wzhuang@fjirsm.ac.cn
猜你喜欢
化工设计通讯(2022年12期)2023-01-24
化工学报(2021年10期)2021-10-31
炼油与化工(2021年3期)2021-07-06
石油化工技术与经济(2020年4期)2020-09-15
化工管理(2020年19期)2020-07-28
石油化工技术与经济(2019年1期)2019-02-14
天然气技术与经济(2018年1期)2018-03-06
自动化博览(2017年2期)2017-06-05
化工设计通讯(2017年6期)2017-03-02
现代工业经济和信息化(2016年6期)2016-05-17
 CHINESE JOURNAL OF CHEMICAL PHYSICS2016年4期
CHINESE JOURNAL OF CHEMICAL PHYSICS2016年4期
- CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- Catalytic Transformation of Oxygenated Organic Compounds into Pure Hydrogen
- Effects of Activation Atmospheres on Structure and Activity of Mo-based Catalyst for Synthesis of Higher Alcohols
- Multiple Plasmonic Resonances and Cascade Effect in Asymmetrical Ag Nanowire Homotrimer
- Depolymerization of Organosolv Lignin over Silica-alumina Catalysts
- Construction and Evaluation of Merged Pharmacophore Based on Peroxisome Proliferator Receptor-Alpha Agonists
- Electricity Storage With High Roundtrip Efficiency in a Reversible Solid Oxide Cell Stack