
Zn高效促进由2-甲酰基苯甲酸合成3-烃基苯酞
2016-10-10 02:39谢国豪周安西廖国富
西北大学学报(自然科学版) 2016年2期
谢国豪,周安西,胡 昕, 廖国富
(上饶师范学院 江西省普通高校应用有机化学重点实验室, 江西 上饶 334001)
·化学与化学工程·
Zn高效促进由2-甲酰基苯甲酸合成3-烃基苯酞
谢国豪,周安西,胡昕, 廖国富
(上饶师范学院 江西省普通高校应用有机化学重点实验室, 江西 上饶334001)
在四氢呋喃(THF)中,Zn高效促进2-甲酰基苯甲酸与溴代烃在35℃下反应,高产率得到相应的3-烃基苯酞。该合成方法条件温和, 操作简单, 原料易得, 产率较高, 为3-烃基苯酞的合成提供了一种新的实用方法。 产物结构经1H NMR,13C NMR, 和HRMS分析表征。
2-甲酰基苯甲酸; 锌; 3-烃基苯酞; 合成
3-烃基苯酞类化合物广泛存在于伞形科植物及其他一些中草药中,具有良好的生物活性[1],部分化合物已经被用于临床。因此,3-烃基苯酞类化合物因其具有广泛的药理活性而受到化学工作者的广泛关注。到目前为止,关于3-烃基苯酞类化合物的合成已有诸多报道[2-14]。但是,通过金属促进合成3-烃基苯酞类化合物的方法却鲜有报道[15-16]。1999年,Wang和本课题组的Xie等人通过金属Sn促进3-羟基苯酞和烯丙基溴反应成功合成得到3-烯丙基苯酞[17-18]。为了进一步丰富3-烃基苯酞的合成方法,利用更为廉价的锌粉来促进2-甲酰基苯甲酸和溴代烃反应成功合成得到3-烃基苯酞(图1)。
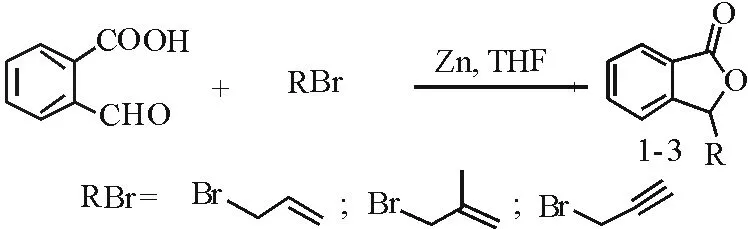
图1 化合物1-3的合成Fig.1 Synthesis of compound 1-3
此外,需要注意的是,由于丁基苯酞较差的水溶性也在一定程度上限制了它在临床上的应用范围。Deng等人发现3-丁基苯酞在体内主要代谢物为羟基衍生物[19]。因此,3-烃基苯酞的羟基衍生物引入了亲水性羟基,可增加药物分子的亲水性,有利于原型药在体内的吸收和代谢。另外,研究表明,一些从天然产物中提取出来的含3-羟基的苯酞类化合物具有良好的药理作用,而且这类化合物还可以作为合成一些杂环化合物的中间体。因此,研究这些羟基衍生物对于寻找这类药用植物的有效成分具有很重要的价值[20]。为此,本课题组在合成3-烯丙基苯酞的基础上进行衍生化,成功得到3-烃基苯酞的羟基衍生物(图2)。
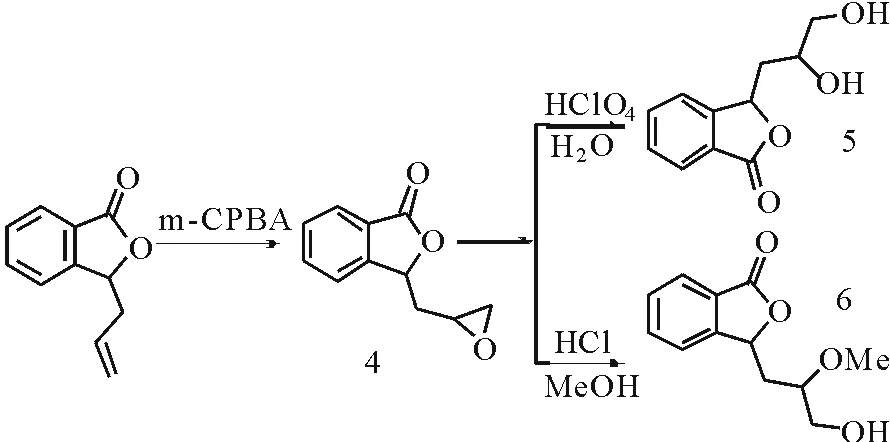
图2 化合物5和6的合成Fig.2 Synthesis of compound 5 and 6
1 实验部分
1.1主要实验仪器与试剂
X-6精密显微熔点测定仪(北京亚力恩机电技术研究院,温度计未校正),Bruker Avance 核磁共振波谱仪(德国Bruker公司)。2-甲酰苯甲酸,锌粉,间氯过氧苯甲酸,高氯酸,烯丙基溴,炔丙基溴,3-溴-2-甲基丙烯等均为阿拉丁试剂公司;甲醇,四氢呋喃,盐酸,石油醚,乙酸乙酯等均为市售分析纯;普通柱层析硅胶(0.058~0.075mm)。
1.23-烯丙基异苯并呋喃-1(3H)-酮(1)的合成
在50mL圆底烧瓶中依次加入邻甲酰苯甲酸(1.06g),锌粉(0.55g),四氢呋喃(10.00mL),最后在室温条件下缓慢滴加烯丙基溴(1.00g)至反应瓶中。滴加完毕后,在35℃条件下反应3h。待TLC检测反应至完全后,加入5mL蒸馏水淬灭反应,然后将反应混合物进行抽滤,并用乙醚反复洗涤残留固体,减压浓缩滤液,出现黄色油状物。将反应也转入分液漏斗中,分出有机相,无机层用乙醚反复萃取至TLC跟踪无产物,合并有机相,用蒸馏水洗涤(15mL×3),再用15mL饱和氯化钠溶液洗涤,最后用无水硫酸钠干燥。减压浓缩,得浅黄色油状物。经硅胶柱层析分离得到浅黄色油状物1.11g,产率为91%。
1H NMR (400 MHz, Chloroform-d)δ7.89 (dd,J=7.6, 1.1 Hz, 1H), 7.68 (td,J=7.5, 1.1 Hz, 1H), 7.56-7.46 (m, 2H), 5.76 (ddt,J=17.1, 10.2, 7.0 Hz, 1H), 5.53 (t,J=5.9 Hz, 1H), 5.22-5.12 (m, 2H), 2.82-2.70 (m, 1H), 2.65 (dtt,J=14.6, 6.5, 1.5 Hz, 1H)。13C NMR (101 MHz, Chloroform-d)δ170.2, 149.2, 133.8, 131.1, 129.1, 126.1, 125.5, 125.5, 121.9, 119.5, 80.1, 77.3, 77.0, 76.7, 38.5。
1.33-(2-甲基烯丙基)异苯并呋喃-1(3H)-酮(2)的合成
在50mL圆底烧瓶中依次加入邻甲酰苯甲酸(1.06g),锌粉(0.55g),四氢呋喃(10mL),最后在室温条件下缓慢滴加2-甲基烯丙基溴(1.14g)至反应瓶中。滴加完毕后,在35℃条件下反应3h。待TLC检测反应至完全后,加入5mL蒸馏水淬灭反应,后处理与化合物1的相同。减压浓缩,得淡黄色油状物。经硅胶柱层析分离得到淡黄色油状物1.17g,产率为89%。
1H NMR (400 MHz, Chloroform-d)δ8.00-7.84 (m, 1H), 7.67 (td,J=7.5, 1.1 Hz, 1H), 7.57-7.42 (m, 2H), 5.61 (dd,J=7.6, 5.6 Hz, 1H), 5.02-4.79 (m, 2H), 2.68-2.49 (m, 2H), 1.85 (s, 3H).13C NMR (101 MHz, Chloroform-d)δ170.3, 149.7, 139.8, 133.8, 129.1, 126.0, 125.6, 122.1, 114.7, 79.5, 42.8, 22.9。
1.43-炔丙基异苯并呋喃-1(3H)-酮(3)的合成
在50mL圆底烧瓶中依次加入邻甲酰苯甲酸(1.06g),锌粉(0.55g),四氢呋喃(10.00mL),最后在室温条件下缓慢滴加炔丙基溴(1.00g)至反应瓶中。滴加完毕后,在35℃条件下反应3h,TLC检测反应至完全。待反应完全后,加入5mL蒸馏水淬灭反应,后处理与化合物1的相同。减压浓缩,得白色固体。经硅胶柱层析分离得到白色固体0.99g,产率为82%。
m.p. 91.6-93.2℃;1H NMR (400 MHz, Chloroform-d)δ7.91 (d,J=7.6 Hz, 1H), 7.78-7.65 (m, 2H), 7.57 (ddd,J=8.1, 5.8, 2.5 Hz, 1H), 5.57 (dd,J=7.3, 4.9 Hz, 1H), 2.97 (ddd,J=16.8, 4.9, 2.7 Hz, 1H), 2.76 (ddd,J=16.7, 7.3, 2.6 Hz, 1H), 2.06 (t,J=2.7 Hz, 1H).13C NMR (101 MHz, Chloroform-d)δ169.7, 148.5, 134.0, 129.6, 126.0, 125.6, 122.4, 77.7, 77.5, 72.0, 24.8。
1.53-(2,3-环氧丙基)异苯并呋喃-1(3H)-酮(4)的合成[17]
称取3-烯丙基苯酞(2.02g)溶于二氯甲烷(40mL)中,在冰水浴冷却下,分批加入间氯过氧苯甲酸(4.28g)(75%,质量分数)。待投料完毕后在此温度下继续反应2h,而后升高至室温条件下反应过夜。待反应完全后,减压浓缩反应液得白色固体,然后加入30mL乙醚将白色固体溶解,并将其转移至分液漏斗中,依次用饱和碳酸氢钠溶液(40mL×4)洗涤,蒸馏水(40mL×3)洗涤后,醚层用无水硫酸钠干燥。浓缩后用硅胶柱层析,浓缩的淡黄色油状物1.95g,产率88%。
dr=1.4;1H NMR (400 MHz, Chloroform-d)δ7.92 (d,J=7.8 Hz, 1H), 7.79-7.65 (m, 1H), 7.63-7.43 (m, 2H), 5.71-5.67, 5.61-5.57 (m, 共1H), 3.29-3.25, 3.06-3.03 (m, 共1H), 2.89-2.86, 2.83-2.79 (m, 共1H), 2.71-2.69, 2.60-2.57 (m, 共1H), 2.33-2.17 (m, 1H), 2.08-2.03 (m, 1H).13C NMR (101 MHz, Chloroform-d)δ170.1, 170.0, 149.2, 149.0, 134.1, 129.3, 125.8, 125.7, 125.6, 122.0, 121.8, 78.8, 78.0, 48.8, 47.9, 47.4, 46.5, 38.7, 36.8。
1.63-(2,3-羟基丙基)异苯并呋喃-1(3H)-酮(5)的合成
在50mL圆底烧瓶中依次加入化合物(42g),四氢呋喃(10mL),水(10mL)和高氯酸溶液(0.25mL),搅拌12h后,减压旋干四氢呋喃,用乙酸乙酯萃取,有机相用蒸馏水洗涤,再用饱和碳酸氢钠溶液洗至pH值为5~7之间,有机层用无水硫酸钠干燥。浓缩后得到淡黄色油状物,经硅胶柱层析分离得到淡黄色油状物1.97g,产率为90%,。
dr=1.4;1H NMR (400 MHz, Chloroform-d)δ7.90-7.86 (m, 1H), 7.70-7.65 (m, 1H), 7.60-7.47 (m, 2H), 5.82-5.77, 5.72-5.67 (m, 共1H), 4.22-4.09 (m, 1H), 3.77-3.66 (m, 1H), 3.60-3.48 (m, 1H), 2.93 (s, 2H), 2.28-2.14 (m, 1H), 2.07-1.94 (m, 1H).13C NMR (101 MHz, Chloroform-d)δ171.2, 170.8, 170.7, 150.2, 149.9, 134.3, 134.2, 129.3, 129.2, 125.8, 125.7, 125.6, 125.5, 122.1, 121.8, 79.3, 78.5, 69.4, 68.7, 66.7, 66.1, 60.4, 38.7, 38.2. HRMS (ESI): m/z: calcd for C11H13O4[M+H]+: 209.080 8; found: 209.081 3。
1.73-(3羟基2甲氧基丙基)异苯并呋喃-1(3H)-酮(6)的合成
在50mL烧杯中加入甲醇(15.00mL),用6 mol/L盐酸调节pH值为3~4之间备用。在50mL圆底烧瓶中依次加入化合物4(2.00g),上述盐酸甲醇溶液(15.00)mL,置于室温条件下进行反应,反应完全后往反应液中加入饱和碳酸氢钠溶液,调节pH值为5~6,然后用乙醚30mL 进行萃取,合并有机相,然后用蒸馏水进行洗涤,有机相用无水硫酸钠干燥,浓缩得到淡黄色油状物,经硅胶柱层析分离得到淡黄色油状物1.94g,产率为83%。
dr=1.2;1H NMR (400 MHz, Chloroform-d)δ7.92-7.88 (m, 1H), 7.72-7.66 (m, 1H), 7.56-7.47 (m, 2H), 5.82-5.77, 5.73-5.69 (m, 共1H), 4.28-4.22, 4.19-4.12 (m, 共1H), 3.55-3.30(m, 5H), 3.00, 2.89 (s, 共1H), 2.22-1.99 (m, 2H).13C NMR (101 MHz, Chloroform-d)δ170.5, 170.3, 150.3, 149.9, 134.1, 129.2, 129.1, 125.8, 125.7, 122.2, 121.8, 78.7, 78.3, 76.7, 76.2, 67.4, 67.0, 59.1, 59.0, 39.2, 38.2. HRMS (ESI):m/z: calcd for C12H15O4[M+H]+: 223.096 5; found: 223.096 1。
2 结果与讨论
2.12-甲酰基苯甲酸烃基化反应
选用2-甲酰基苯甲酸与烯丙基溴的反应为模型反应,考察Zn粉的促进效果及其他因素对反应的影响。首先,基于Sn促进合成3-烯丙基苯酞的研究,选取n(2-甲酰基苯甲酸)∶n(烯丙基溴)∶n(锌粉)为1∶1.2∶1.2,在THF溶剂中35℃条件下进行反应,以91%的产率得到目标产物。然后,通过单因素优化法对反应温度和反应时间进行了考察。发现降低反应温度时,反应3h后反应不完全;而升高温度至50℃对产物收率影响不大;此外,延长反应时间对反应无明显影响。在确定了反应条件之后,对该反应中卤代烃进行了考察。当卤代烃为2-甲基-3-溴丙烯或炔丙基溴时(见表1),分别以89%和82%的收率得到对应的目标产物。当卤代烃为碘甲烷或氯代烃时,则没有得到对应的目标产物。
表1底物扩展
Tab.1Screen of the substrate
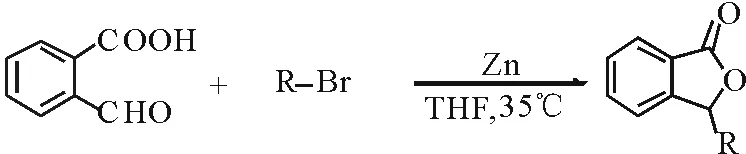

序号RBr产物收率/%191289382
2.23-烃基苯酞的羟基衍生物的合成
3-烃基苯酞的羟基衍生物的合成是经过两个过程完成,首先是经过环氧化反应,用有机过氧酸对烯烃进行环氧化反应时目前较为常见的一种方法,通常情况下,烯烃分子中含有给电子基团,而过氧酸中含有吸电子基团时,反应更容易进行。3-烯丙基苯酞是一个末端烯烃,环氧化反应比非末端烯烃更困难。因此,本文采用适当过量的间氯过氧苯甲酸对3-烯丙基苯酞进行环氧化,确保其双键充分被氧化,得到对应的环氧化合物,从而有利于反应产率的提高和简化后期分离工作的难度。其次是对环氧化合物进行开环反应,无论是水解开环或者是醇解开环,都是在酸性条件下进行,该反应过程特别需要注意pH值的控制;特别是醇解开环的过程,碱性开环与酸性开环所得产物不同。这是因为环氧化合物的开环反应是一种特殊的亲核取代反应。在酸性条件下,环氧化合物首先质子化使碳氧键削弱,将较差的离去基团烷氧负离子转变为较好的离去基团醇。然后,亲核试剂进攻中心碳原子,使碳氧键断裂发生开环[21-22]。在这种亲核取代反应中,反应的主要动力是酸对环氧化合物的质子化,使碳氧键削弱,而使亲核性较弱的亲核试剂也可以顺利进行反应。在碱性条件下,环氧化合物不能质子化,碳氧键比较牢固,离去基团烷氧负离子的离去倾向比较弱,不利于开环反应的进行[23],反应动力来源于强的亲核试剂对中心碳原子的进攻。因此,较强的亲核试剂才能顺利进行反应。另外,碱性条件下碳氧键断裂应在取代基较少的一边,因为空间位阻更小,更有利于亲核试剂的进攻;而酸性条件下断裂连接取代基较多的一侧,因为取代基越多,越有利于正电荷的稳定。
2.3可能的反应机理
该反应可以通过两种途径来实现:其一,与3-羟基苯酞和格氏试剂的反应类似,首先溴代烃与金属锌反应得到有机锌试剂,同时由2-甲酰基苯甲酸得到3-羟基苯酞,而后3-羟基苯肽与有机锌试剂作用从而得到3-烃基苯酞(Path A)[14]。其二,与铟促进2-甲酰基苯甲酸烯丙基化反应相似[16],同样由溴代烃与金属锌反应得到有机锌试剂,然后与2-甲酰基苯甲酸反应,经过亲核加成、分子内酯化反应,最后离去Zn(OH)Br,从而得到3-烃基苯酞(Path B)。

图3 可能的反应机理Fig.3 Proposed mechanism
3 结 语
建立了一种Zn促进的由2-甲酰基苯甲酸与溴代烃反应高效制备3-烃基苯酞的方法。与传统制备3-烃基苯酞的方法相比,该方法不需要在无水无氧条件下进行且本法具有反应时间短、反应条件温和、后处理简单、产物产率高等特点。此外,对3-烯丙基苯酞进行衍生化反应:通过环氧化反应、水解或醇解开环反应,从而成功获得相对应的3-烃基苯酞羟基衍生物。
[1]MATSUDA H, SHIMODA H, YOSHIKAWA M. Structure-requirements of isocoumarins, phthalides, and stilbenes from hydrangeae dulcis folium for inhibitory activity on histamine release from rat peritoneal mast cells[J]. Bioorganic & Medicinal Chem. 1999, 7(7): 1445-1450.
[2]李绍白. (±)-3-正丁基-4-甲氧基苯酞的合成[J].合成化学, 1993, 1(3): 255-257.
[3]李绍白, 王志伟.(±)-3-正丁基-5-甲氧基苯酞的合成[J].兰州大学学报(自然科学版), 1997, 33(2): 120-122.
[4]PALEO M R, LAMAS C, CASTEDO L, et al. A new synthesis of phthalides by internal trapping in ortho-lithiated carbamates derived from benzylic alcohols [J].J Org Chem, 1992,57(7):2029-2033.
[5]KURIYAMA M, ISHIYAMA N, SHIMAZAWA R, et al. Efficient synthesis of 3-arylphthalides using palladium-catalyzed arylation of aldehydes with organoboronic acids[J].J Org Chem,2009, 74(23):9210-9213.
[6]GARIBAY P, VEDSO P, BEGTRUP M, et al. Solid-phase directed ortho-lithiation and the preparation of a phthalide library[J].J Comb Chem,2001, 3(4): 332-340.
[7]MUKHOPADHYAY R, KUNDU N G. A convenient and highly regio and stereoselective method for the synthesis of (E)-3-alkylidene isobenzofuran-1(3H)-ones (phthalides)[J]. Tetrahedron,2001, 57(46): 9475-9480.
[8]HAYAT S, RAHMAN A U, CHOUDHARY M I, et al. An improved method for the synthesis ofγ-lactones using sodium bromate and sodium hydrogen sulfite[J].Tetrahedron Lett,2001, 42(9):1647-1649.
[9]SAKAMOTO M, SEKINE N, MIYOSHI H, et al. Absolute asymmetric phthalide synthesis via the solid-state photoreaction of N,N-disubstituted 2-benzoylbenzamides involving a radical pair intermediate [J].J Am Chem Soc,2000, 122(41): 10210-10211.
[10] JAYARMAN M, FANWICK P E, CUSHMAN M. Novel oxidative transformation of indenoisoquinolines to Isoquinoline-3-spiro-3′-phthalides in the presence of osmium tetraoxide and 4-methylmorpholine N-oxide[J].J Org Chem,1998,63(17):5736-5737.
[11] CIATTINI P G, MASTROPIETRO G, MORERA E,et al. A new synthesis of 3-ylidenephthalides via palladium-catalyzed cyclocarbonylation of 2-triflyloxyacetophenones[J].Tetrahedron Lett, 1993, 34(23): 3763-3766.
[12] MORI S, TAKECHI S, SHIMIZU S, et al. Convergent synthesis of S-8921, a new potent hypocholesterolemic arylnaphthalene lignan analog[J]. Tetrahedron Lett, 1999, 40(6): 1165-1168.
[13] MALI R S, BABU K N. Reactions of 3-(1-hydroxyalkyl)phthalides with acids:Synthesis of (Z)-3-alkylidenephthalides and 3-alkyl-8-hydroxyisocoumarins[J].J Org Chem, 1998, 63(8):2488-2492.
[14] CANONNE P, PLMAONDON I, AKSSIRA I. Reactions selectives des organomagnesiens avec les lactols et les lactones synthese des diols primaires-secondaires[J].Terrahedron,1988, 44(10): 2903-2912.
[15] LEE J H, PARK Y S, NAM M H, et al. Indium-mediated allylation of hydroxyphthalides with acetic acid as an additive[J].Bull Korean Chem Soc, 2005, 26(3): 496-498.
[16] MIRABDOLBAGHI R, DUDDING T. An indium-mediated allylative/transesterification DFT-directed approach to chiral C(3)-functionalized phthalides[J].Org Lett, 2012, 14(14): 3748-3751.
[17] WANG G X, XIE G H, WU Y L, et al. The synthesis of 3-(2-hydroxybutyl)isobenzofuran-1(3H)-one[J]. Chin Chem Letters, 1999, 10(1): 21-22.
[18] 王国新, 谢国豪, 吴元鎏, 等. 3-(3′-羟基丁基)异苯并呋喃-1(3H)-酮的合成[J]. 合成化学, 2000, 8(1): 63-66.
[19] DENG W B, FENG Y P. Effect of dl-3-butylphthalide on brain edema in rats subjected to focal cerebral ischemia[J]. Chin Med Sci J, 1997, 12(1):102-106.
[20] ZHANG Y, WANG L, ZHANG L Y, et al. Effects of 2-(1-hydroxypentyl)-benzoate on platelet aggregation and thrombus formation in rats[J]. Drug Development Research, 2004, 63(4): 174-180.
[21] WANG L, LI Y, HUANG H, et al. A novel synthesis of the monobactam antibiotic carumonarn[J].Chin J Org Chem, 2006, 26:1208-1216.
[22] 林军, 潘圣强, 张从海, 等. 手性β-氨基醇的合成及其在不对称催化中的应用进展[J].云南民族大学学报(自然科学版), 2010, 19(1):1-9.
[23] 翁文, 周宏英, 傅宏祥. 手性氨基醇在不对称催化中的应用及新进展[J]. 有机化学, 1998, 18(6): 509-520.
(编辑陈镱文)
An efficient Zn-mediated synthesis of 3-alkyl phthalide from 2-formylbenzoic acid
XIE Guo-hao, ZHOU An-xi, HU Xin, LIAO Guo-fu
(Key Laboratory of Applied Organic Chemistry, Higher Institutions of Jiangxi Province,Shangrao Normal University, Shangrao 334001, China)
A simple and efficient method for the conversion of 2-formylbenzoic acid to the corresponding 3-Alkyl Phthalide by using Zinc in THF was developed. The method proposed here has multiple advantages, such as mild condition, easy procedure, cheap ingredients and high yield, which provide a new and practical protocol for the synthesis of 3-Aliphatic Phthalide. The structures of products were characterized by1H NMR,13C NMR, and HRMS.
2-formylbenzoic acid; zinc; 3-alkyl phthalide; synthesis
2015-04-11
江西省教育厅科技基金资助项目(GJJ09642)
谢国豪,男,江西上饶人,上饶师范学院教授,从事药物合成研究。
O261.256.4
A
10.16152/j.cnki.xdxbzr.2016-02-012
猜你喜欢
中国调味品(2022年12期)2022-12-05
林产化学与工业(2021年2期)2021-05-11
吉林农业(2019年6期)2019-06-11
无机化学学报(2019年2期)2019-02-27
教育教学论坛(2018年38期)2018-09-25
中成药(2017年10期)2017-11-16
中成药(2017年4期)2017-05-17
中国洗涤用品工业(2017年2期)2017-04-16
中国洗涤用品工业(2016年2期)2016-02-28
烟草科技(2015年8期)2015-12-20
