
有氧运动对NAFLD大鼠肝细胞线粒体结构功能的影响
2017-01-04 01:08刘倩倩侯改霞习雪峰肖国强
中国体育科技 2016年5期
刘倩倩,秦 智,侯改霞,习雪峰,肖国强
有氧运动对NAFLD大鼠肝细胞线粒体结构功能的影响
刘倩倩1,秦 智2,侯改霞1,习雪峰1,肖国强3
目的:观察8周60 min/天的游泳训练对NAFLD大鼠肝脏脂质异常蓄积、细胞线粒体结构和功能紊乱的改善作用。方法:3月龄SPF级雄性SD大鼠35只,随机分为正常对照(C组,9只),标准饲料喂食;高脂组(26只),高脂饲料喂食,构建NAFLD模型:分为高脂对照组(FC组,13只)和高脂运动组(FE组,13只)。采用无负重游泳训练,每周运动6天,休息1天,持续8周。运动结束后,测定肝指数、血脂水平、肝组织切片、电镜观察肝细胞线粒体结构,ORT-PCR检测细胞因子信号转导抑制物3(SOCS3)、固醇调节元件结合蛋白1c(SREBP1c)等mRNA水平。结果:与C组相比,FC组大鼠体质量、肝质量、肝脂数、TC、FFA、SREBP1c、SOCS3 mRNA表达均出现明显增加(P<0.01或P<0.05),HDL-c减少(P<0.05);FE组与FC组相比,体质量、肝脏质量、肝指数、TC、FFA、SREBP1c、SOCS3 mRNA表达均出现显著下降(P<0.01或P<0.05);镜下显示,C组大鼠肝结构清晰,细胞内无脂肪小泡,线粒体嵴丰富齐整;FC组大鼠肝结构紊乱,核畸形,核周水肿,线粒体肿胀,嵴部分排列紊乱或消失,胞内脂滴及空泡样变数量明显增多;FE组大鼠肝结构改善,膜结构正常,线粒体内嵴结构较为明显。结论:1)16周高脂饮食NAFLD大鼠肝脏出现了明显的肝脏脂质异常蓄积,肝组织结构紊乱,肝细胞内线粒体数量减少,结构畸形。2)规律的适宜强度的有氧运动可明显改善NAFLD大鼠中存在的脂质代谢紊乱,可能通过促进肝脏细胞内线粒体的数量、形态结构、功能趋于正常,从而减少肝脏这一非脂肪组织的脂肪蓄积,减轻肝脏脂肪变性程度,促进非酒精性脂肪肝的良性转归。关键词:非酒精性脂肪肝;非脂肪组织;肝细胞;线粒体结构;脂肪变性;大鼠
非酒精性脂肪肝(nonalcoholic fatty liver disease,NAFLD)是脂肪在肝脏这一非脂肪组织异常蓄积并引起肝组织连续应激的过程,病谱包括非酒精性单纯性脂肪肝、非酒精性脂肪性肝炎、肝硬化和肝癌,其发病率呈增高[12,27]和低龄化趋势[10,19],NAFLD已成为一个世界性的健康问题[14]。肝细胞线粒体功能障碍在NAFLD的发病过程中起着重要作用[7],线粒体是肝细胞最重要的产能细胞器,它在细胞能量产生过程中扮演重要角色,并整合多个信号途径来控制细胞的生命和死亡[17]。线粒体功能障碍使肝脏对脂质异常蓄积这一有害刺激耐受性降低,继而发生肝组织脂肪变性,肝脏脂肪变性又反作用于线粒体[13,17],引起线粒体合成ATP的能力降低,超微结构发生改变,同时诱导肝细胞损伤信号通路作用,形成一个恶性循环。膳食结构的良性调整在预防NAFLD的发生发展中起重要作用,部分是通过维持线粒体的功能达到预防效果[9]。目前,有关于运动改善NAFLD中线粒体功能的研究较少,其具体机制尚不清楚,本研究旨在通过观察运动对NAFLD中肝细胞线粒体的超微结构和相关基因的影响,以期为其防治提供一定的理论基础。
1 实验对象与方法
1.1 实验动物与饲料
3月龄SPF级雄性Sprague-Dawley (SD)大鼠35只,体质量200±22 g,由广州中医药大学实验动物中心提供,许可证号:SCXK(粤)2008-0002粤监证字2008D007。常规分笼喂养,自然光照,饲养环境温度23℃±2℃,自由饮水进食。
普通饲料:选用国家标准啮齿类动物混合饲料,由广州中医药大学实验动物中心提供。高脂饲料:采购于广东省医学实验动物中心。高脂饲料具体成分:5%(w/w)蔗糖,18%(w/w)猪油,15%(w/w)蛋黄粉,0.5%(w/w)胆盐,1%(w/w)胆固醇,60.5%(w/w)基础饲料。
1.2 动物分组与模型构建
适应性喂养后,随机抽取10只大鼠,喂食普通饲料,作为正常对照组(C组);剩余30只喂食高脂高胆固醇饲料,用于制备大鼠NAFLD模型。分组后,正常对照组大鼠喂食普通饲料,高脂组大鼠喂食高脂饲料。两组大鼠均不限制饮食和饮水。喂食8周,每周同一时间记录大鼠体重,及时剔除体重显著性差异的个体,保持同组别个体间体重均无显著性差异。于造模期的第6、7、8周两组均宰杀1只大鼠,制备肝组织切片和测生化指标,确定模型进度,直至建模成功。建模成功后,高脂模型组大鼠分为高脂对照组(FC组)和高脂运动组(FE组)。
依据中华医学会肝脏病学分会脂肪肝和酒精性肝病学组颁布的非酒精性脂肪肝病诊断标准[16],控制本研究动物造模质量。本研究SD大鼠NAFLD模型诊断标准为:1)血清ALT基本正常;2)肝脏组织学表现符合非酒精性脂肪肝诊断标准:低倍镜下视野内占肝细胞总数30%以上的肝细胞发生脂肪变性,但无其他明显组织学改变。视野内30%~50%的肝细胞脂肪变者为轻度脂肪肝;50%~75%为中度脂肪肝;75%以上为重度脂肪肝。低倍镜下视野内发生脂肪变的肝细胞占肝细胞总数的比例<30%者称为肝细胞脂肪变性。肝组织病理学切片送两位执业病理医师盲法诊断。
1.3 运动方案
高脂运动组大鼠自运动周期开始分别进行为期8周的不负重游泳训练,第1周为适应性训练。第1天运动时间为10 min,以后每天递增10 min,直至游泳时间增加到60 min,从第2周开始维持此运动时间至9周结束。
1.4 取材
运动周期实验结束后,禁食12 h,大鼠腹腔注射10%水合氯醛(剂量:0.35~0.40 mL/kg bw)麻醉,腹主动脉取血,静置、处理,用于血脂指标测试。在大鼠尚存心跳时,迅速取肝,处理大小为1~2 mm3,1 min之内入2.5%~3%戊二醛,4℃保存2~4 h。取大鼠肝脏组织置于10%中性甲醛溶液固定24~48 h,用于制备肝组织病理切片;取部分肝组织,放入液氮保存、待测。
1.5 检测指标及方法
1)整个实验期间,每周固定时间称量大鼠体质量;2)采用半自动生化分析仪Eppendorf ECOM-F 6124(Eppendorf Co.Germany)检测血清中丙氨酸转氨酶(ALT)、总胆固醇(TC)、甘油三酯 (TG)、低密度脂蛋白胆固醇(LDL-C)以及高密度脂蛋白胆固醇(HDL-C)含量;3)血清游离脂肪酸(free fatty acid,FFA)采用比色法;4)肝指数:称量肝重与体质量,计算肝/体比值,即为肝指数;5)通过HE染色制作肝组织病理切片,光镜下观察大鼠肝脏脂肪变性情况;6)肝组织电镜切片,通过日立H-7650透射电子显微镜观察线粒体结构;7)信使核糖核酸测定:实时荧光定量检测细胞因子信号转导抑制物3(suppressor of cytokine signaling 3,SOCS3)、固醇调节元件结合蛋白1c(sterol regulatory element binding protein 1c,SREBP1c)、过氧化物酶体增殖物激活受体α(peroxisome proliferator activated receptor α,PPARα)、过氧化物酶体增殖物激活受体γ辅助激活因子1(peroxisome proliferator activated receptor-γ coactivator-1,PGC-1 )的表达,GAPDH作为内参。
1.6 肝组织病理切片
常规石蜡切片。HE染色,光镜下观测肝组织学变化。将组织块选取同一部位进行切块并修块、脱水、冲水透明后,浸蜡、包埋、切片、捞片、摊片,在60℃干燥箱中过夜后,进行HE染色。
1.7 RT-PCR检测方法
取材后,立即进行RNA的制备。加入1 mL的Trizol提取总RNA。准备 1.5 mL 的 EP管,冰上操作:在 EP管中加入0.1~5 μg模板 RNA,1 μL Oligo(dT)18引物, DEPC水定容至12 μL,短暂离心;70℃水浴5 min后,冰上冷冻;短暂离心后,冰上操作:加入 4 μL 5×First-Strand Buffer,1 μL RibolockTM Ribonuclease Inhibitor,2 μL(10 mmol)dNTP mix;短暂离心,37℃水浴5 min,加入1 μL (200 U/μL)ReverAid M-MuLV反转录酶(总反应体积20 μL); 42℃水浴60 min进行反转录;70℃水浴10 min灭活逆转录酶,终止反应,再冰上冷冻;立即进行qRT-PCR。 qRT-PCR反应条件为:95℃预变性2 min;95℃变性15 s,65℃退火15 s,循环40次。试剂购于威佳生物试剂公司。目的基因引物序列如表1所示。GAPDH为内参,根据公式2-△△Ct计算目的基因相对表达量。
1.8 肝组织透射电镜方法
取新鲜肝组织,大小为1~2 mm3,取材的刀片要锋利,低温操作,通常为4℃;前固定:1 min之内入2.5%~3%戊二醛,4℃,2~4 h。0.1 mol/L PBS漂洗,10 min×2;后固定:1%锇酸1.5~2 h,0.1 mol/L PBS 漂洗,5 min×2。脱水:50%、70%乙醇逐级脱水,每级10 min,80%、90%、100%丙酮逐级脱水,每级10 min×2。置换:100%丙酮,环氧树脂Epon812包埋剂以1∶1混合,置换40 min。浸透:环氧树脂Epon812包埋剂37℃浸透过夜。包埋:将组织置包埋模内Epon812包埋剂包埋,60℃聚合48 h。在解剖显微镜下修块,将组织表面修平整,暴露组织并根据组织表面的形状修成梯形、正方形或长方形。切半薄切片:在AO超薄切片机下切1 μm的半薄切片,并置水上展平。HE染色。在解剖显微镜下和普通显微镜下定位,通常定位后切面为0.09 mm3;超薄切片:在AO超薄切片机下切40~60 nm的超薄切片,用铜网捞片。电子染色:70%乙醇配制的饱和醋酸铀染色3 min,双蒸水漂洗,铅染液染3 min,双蒸水漂洗。干燥后电镜观察。

表 1 本研究所涉全部QRT-PCR引物序列
1.9 统计方法

2 实验结果与分析
2.1 造模期正常对照组和高脂组大鼠体重的变化
在造模周期内,正常对照组和高脂组SD大鼠的体重均有明显增加,从适应性喂养的300 g左右增加到造模周期结束时的580 g左右,但两组大鼠体重增加的幅度并不一致,具体数据如表2。
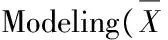
表 2 造模期间大鼠体重变化
注:与正常对照组大鼠适就周比较,##表示P<0.01;与高脂组大鼠适应周比较,**表示P<0.01;与正常对照组对应周比较,§ 表示P<0.05,§§表示P<0.01。
在适应性喂养的1周内,两组大鼠体重无明显差异(P>0.05),进入造模周期后,两组大鼠体重较适应周均有明显增长,从造模第1周就呈非常著性差异(P<0.01),此趋势一直持续至建模期第8周。同时,造模期第1周,高脂组大鼠体重与正常对照组相比,呈显著性差异(P<0.05);第2、3周,两组大鼠体重又趋于相近,差异不具统计学意义;从第4周开始至建模第8周,高脂组大鼠体重均高于同期普通正常对照组大鼠体重,且差异具有非常显著性(P<0.01)。
2.2 各组大鼠肝指数的改变
表3显示,运动周期结束后,FC大鼠组与C组比较,体质量平均增加22%(P<0.01),肝脏质量平均增加74%(P<0.01),肝指数增加42%(P<0.01);FE组大鼠与C组相比,体质量略有下降,但无显著差异(P>0.05),肝脏质量增加25%(P<0.01),肝指数增加30%(P<0.01);FE组大鼠与FC组相比,体质量下降26%(P<0.01),肝脏质量下降28%(P<0.01),肝指数下降8%(P<0.01)。
2.3 各组大鼠血清脂代谢指标的变化
实验周期结束后,FC组大鼠血清TC明显高于C组,差异具有非常显著性(P<0.01);FE组大鼠TC水平与C组相比,有所升高,差异具有显著性(P<0.05),但与FC组相比,有所降低,差异具有显著性(P<0.05);TG水平,FC组大鼠明显高于C组和FE组,差异具有非常显著性(P<0.01),FE组与C组相比,略有上升,差异不具有显著性(P>0.05);FC组大鼠HDL-c与C组相比,有所下降(P>0.05);FE组大鼠HDL-c与C组相比降低明显,差异具有显著性(P<0.05);FC组和FE组大鼠LDL-c与C组相比,均有所上升,其中,FC组增加幅度较小(P>0.05),FE组增高较多,差异具有显著性(P<0.05);FC组大鼠FFA水平与C组相比,平均增加了370%(P<0.01),FE组大鼠与C组相比,FFA水平平均增加211%(P<0.01),FE组大鼠与FC组相比,FFA水平平均下降43%(P<0.01,表4)。
表 3 各组大鼠体质量、肝脏质量及肝指数变化


n体质量(g)肝脏质量(g)肝指数(×10-3)C组7581.75±61.60 12.52±1.57 21.54±1.87 FC组7713.13±44.33☆☆21.87±3.17☆☆30.61±3.43☆☆FE组7562.00±48.89§§15.72±3.55☆☆§§28.02±5.78☆☆§§
注:与C组相比较,☆☆表示P<0.01;与FC组相比较,§§表示P<0.01,下同。

表 4 各组大鼠血清脂代谢各指标变化
注:与C组相比较,☆表示P<0.05,☆☆表示P<0.01;与FC组相比较,§表示P<0.05,§§表示P<0.01,。
2.4 肝组织病理学改变
通过肉眼观察,正常对照组大鼠肝脏组织多呈暗红色,边缘锐利,光镜下如图1a所示,肝小叶结构清晰,肝条索排列整齐,肝细胞内无脂肪小泡;高脂对照组肉眼观察肝组织体积增大,边缘圆钝,颜色发黄,切面有油腻感,光镜如图1b所示,镜下多见肝细胞肿胀、气球样变,胞质内脂滴沉积,肝小叶、肝条索不清晰;高脂运动组肝组织颜色肉眼观察接近于正常对照组,光镜下见图1c,肝小叶、肝条索状结构尚清晰,少量脂肪空泡,无明显炎性细胞浸润。

图 1 各组大鼠肝组织病理学变化示意图
2.5 肝组织超微结构的改变
正常对照组大鼠肝组织电镜显示核大而圆,居细胞一侧,核膜清晰,线粒体、内质网等细胞器密集丰富,膜结构正常,形态良好,线粒体嵴丰富齐整;内质网发达,糖原丰富,溶酶体少见(图2)。高脂对照组大鼠电镜下表现为核畸形,核周水肿,线粒体肿胀,嵴部分排列紊乱或消失,细胞内脂滴及空泡样变数量明显增多,粗面内质网上核糖体有脱落现象(图3)。高脂运动组大鼠镜下可见膜结构正常,胞质内线粒体及内质网较为密集,细胞内可见脂滴及空泡样变,线粒体内嵴结构较为明显,有胶原纤维沉积,与高脂对照组相比高脂运动组大鼠核畸形有所减轻,细胞内脂滴及空泡样变数量较高脂组明显减少(图4)。

图 2 正常对照组大鼠肝脏超微结构示意图
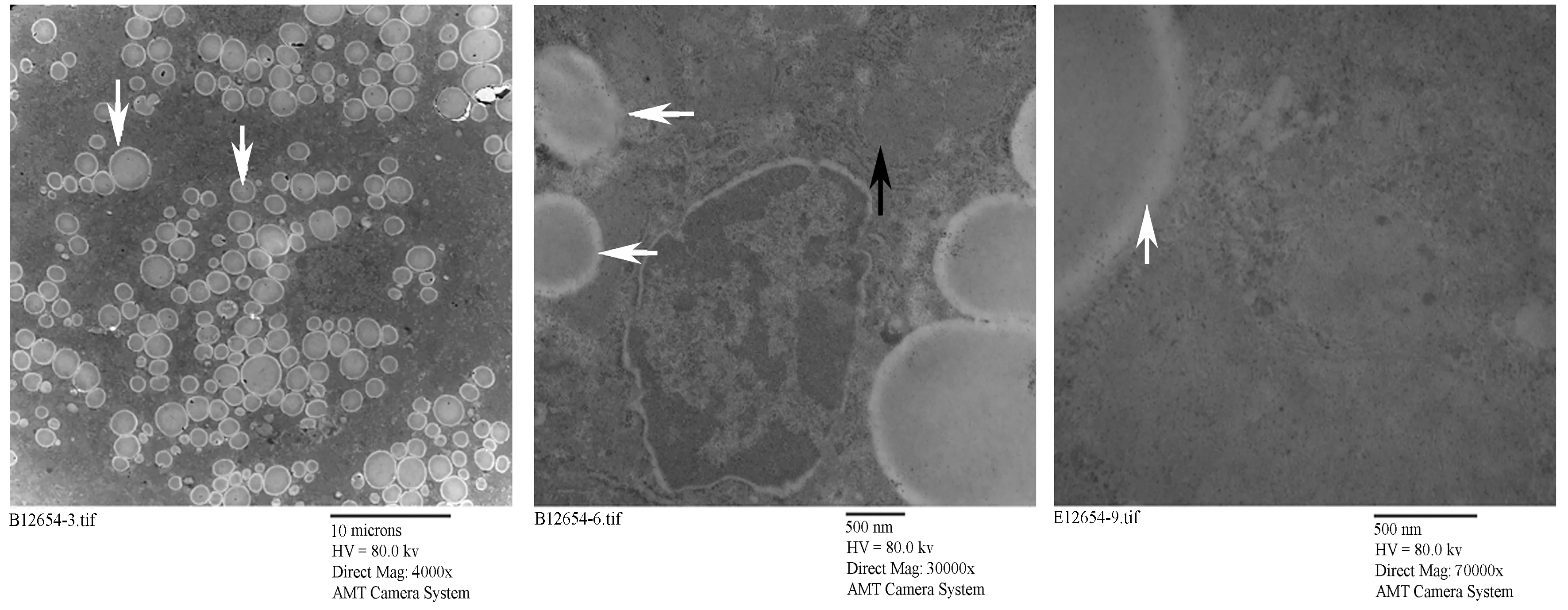
图 3 高脂对照组大鼠肝脏超微结构示意图
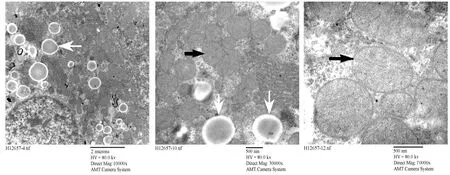
图 4 高脂运动组大鼠肝脏超微结构示意图
2.6 各组大鼠肝脏线粒体相关基因的变化
表5结果显示,与C组比较,FC、FE组SREBP1c、SOCS3 mRNA表达水平增加明显,具有非常显著性差异(P<0.05), FE组与FC组相比,SREBP1c、SOCS3 mRNA表达水平降低明显,差异具有非常显著性(P<0.05);与C组相比,FC组PPARα mRNA表达水平有所增加,但差异不具有统计学意义(P>0.05),FE组组内个体差异较大,与C组相比,PPARα mRNA表达水平差异不明显(P>0.05);与C组相比,FC组和FE组PGC1α mRNA表达水平有所下降,但差异不具有统计学意义(P>0.05)。
表 5 各组大鼠肝脏线粒体相关基因mRNA表达变化
3 讨论与分析
3.1 高脂饮食和运动对大鼠肝脏质量和血脂代谢的影响
肝脏是人体内最大的代谢器官,在调节糖、脂代谢中起着非常重要的作用。NAFLD的发病机制至今尚不完全明确,有多种假说,其中为多数学者接受的是“多平行打击学说”和“二次打击学说”。Tilg等提出了“多平行打击学说”[21]。该学说强调了一些非常不同的并行过程导致了肝脏炎症地发生——特别是肝外组织如肠和(或)脂肪组织促进了肝脏炎症的发生发展,且肝脏炎症可能先于脂肪肝、肝硬化和肝癌,或者说在一定比例上先于它们发生。Day等第一次将NAFLD和胰岛素抵抗(Insulin Resistance,IR)联系在一起,提出了“二次打击学说”[11]。其中,第1次打击先是甘油三酯累积,脂肪变性,系统性IR的结果,第2次打击被认为是由于甘油三酯的长期累积,导致肝脏氧化应激。这种应激会导致谷胱甘肽和氧化应激之间的不平衡,线粒体功能受损,脂肪酸β氧化功能受损。自由基和脂质过氧化产物导致肝细胞损伤,直接破坏细胞器或细胞膜,导致细胞坏死或凋亡及介导炎症反应。在此过程中,肝脏持续不断的慢性损伤将导致肝纤维化、肝硬化和肝癌。后续研究显示,人体内高胰岛素血症(第1次打击)确实先于脂肪肝地发展[20],同时第2次打击更为精细。除此之外,也出现了很多其他模型,如“四步”理论强调脂质释放和由于肝静脉阻塞进一步发展为肝硬化[26]。该模型解释疾病形态学变化比较有效,相较于其他关于NAFLD的理论更有助于理解。
在NAFLD患者中多存在糖脂代谢紊乱,体重增加的现象,本实验在造模期间高脂组大鼠体重与正常对照组大鼠体重均出现了持续增长的现像,但增长幅度略有不同,两组大鼠体重在造模第1周开始出现差异,且差异具有显著性;在第2、3周体重差异减小,不具统计学意义;第4周开始体重增长差别明显,差异具有非常显著性,这种状态一直持续到造模的最后一周,第8周。通过高脂饮食构建非酒精性脂肪肝大鼠模型时,高脂组大鼠体重与正常对照组相比较会出现明显增长,体重增长幅度大于正常对照组,提示,高脂饮食可能更易引起机体的脂肪蓄积,这与前人的研究结果一致[1,2]。
饮食诱导的肥胖在实验动物和人群中普遍存在,肥胖和超重也是NAFLD获得认可的致病因素。在运动实验周期结束后,本研究FC组大鼠中TC、TG和LDL-C水平明显上升,表明高脂饮食在短期内即造成大鼠体内脂质代谢紊乱,为SD大鼠构建非酒精性脂肪肝模型提供可能。同时,游离脂肪酸出现了非常明显的升高,差异具有非常显著性,提示,高脂饮食引起大鼠脂质代谢功能障碍,游离脂肪酸不能及时利用和消除,在体内异常蓄积为下一步胰岛素抵抗的恶性循环及加重机体的氧化应激等提供了充分可能。本实验中游离脂肪酸的显著增加即与第1次打击相符,脂质代谢障碍使外周脂肪分解速率加快并伴随有血清游离脂肪酸的增加,不断增加的FFA被送到肝脏,超过了肝脏正常负荷,正常的合成分解动态平衡被破坏,FFA的脂毒性作用于肝脏,使肝脏的氧化利用能力降低,脂质在肝脏堆积,进而参与脂肪肝地形成。这使得本研究的FC组大鼠肝脏质量明显高于C组,FE组虽有一定程度的改善,但仍高于C组。同时,FFA的增多也可以作用于第2次打击,使胰岛素抵抗进一步恶化,加重肝细胞的脂肪变性,形成一个恶性循环,最终发展成为脂肪肝。
3.2 高脂饮食和运动对肝细胞线粒体形态结构的影响
游离脂肪酸FFA不仅是细胞代谢的能源物质,并且在机体调节糖脂代谢和IR中是一种重要的介质,起着非常重要的作用。当体内FFA含量增多,超出生理负荷时,就会有大量的FFA从脂肪细胞中溢出,进而引起脂毒性作用。其具体表现为大量的FFA涌入肝脏和肌肉组织,以甘油三酯的形态在肝细胞内异常蓄积。此外,FFA在抑制外周葡萄糖利用的同时刺激肝脏的糖异生,使血浆FFA增多。肝脏脂肪变性会引起脂质过氧化,诱发产生大量的促氧化物的同时伴随抗氧化物的大量减少,从而促使肝细胞自由基增多,形成一系列的自由基及其代谢产物,如MDA、GSH-PX,4-羟基壬烯酸等,导致肝细胞内蛋白发生交联,形成Mallory小体等[5,29]。肝细胞是体内代谢比较旺盛的器官之一,线粒体含量极丰富,且线粒体又是脂肪酸氧化利用的主要细胞器。FFA的异常增多可以引起线粒体的活性氧自由基ROS即内源性自由基增加,启动不饱和脂肪酸氧化,经过一系列的反应形成脂质过氧化物LPO,LPO负反馈于线粒体并改变其DNA从而抑制电子呼吸链的传递,如此反复,最终导致肝脏脂质过氧化,使肝细胞内的细胞器出现数量减少或结构异常的现象。如肝细胞线粒体数量减少,出现肿胀和通透性增加等结构变化,甚至引起内质网应激进而出现肝细胞脂性凋亡。
本实验显示,C组大鼠肝细胞内细胞器含量丰富、核大、居中央,清晰可见大量的内质网且内质网附着核糖体、线粒体,线粒体排列密集,嵴结构清晰。高脂对照组大鼠出现了典型的单个肝细胞内脂肪小泡密集出现,细胞器3 000倍视野下几乎不可见,高倍镜下内质网大量减少,同时伴随核糖体减少或消失,线粒体数量减少,出现结构肿胀,胞内随处可见增多的溶酶体和脂肪小泡,部分线粒体嵴突大量消失或不见。高脂运动组大鼠线粒体数量和结构有所恢复,内质网、核糖体等结构较高脂对照组也有明显的改善,溶酶体和脂肪小泡可见。脂肪变化可以引起体内器官产生一些相适应的形态学变化,脂肪在肝脏累积形成脂肪肝、脂肪蓄积引起胰腺形态学变化可以更好的标记胰脂毒性。有研究表明胰腺的脂肪变性通过内镜超声可见。有趣的是,引起“脂肪胰腺”的危险因素与脂肪肝的危险因素互相重叠[6,8],提示存在共同的脂毒性作用、脂质异位沉积、组织中额外脂肪蓄积最终导致组织结构破坏和器官功能障碍。
3.3 高脂饮食和运动对肝脏线粒体相关基因表达的影响
有研究[1]发现,NAFLD与血清瘦素水平关系密切,是脂肪肝发生的独立危险因素。这与其他学者如Tobe等[22]的发现相同,多元回归显示,Leptin是脂肪肝的独立危险因素。而SOCS3的高表达是瘦素抵抗的标志[4,25],上调的SOCS3引起肝脏成脂基因SREBP1c的高表达(图5),引起脂质在肝脏这一非脂肪组织异常蓄积,进而引起肝细胞、线粒体结构和功能的异常。
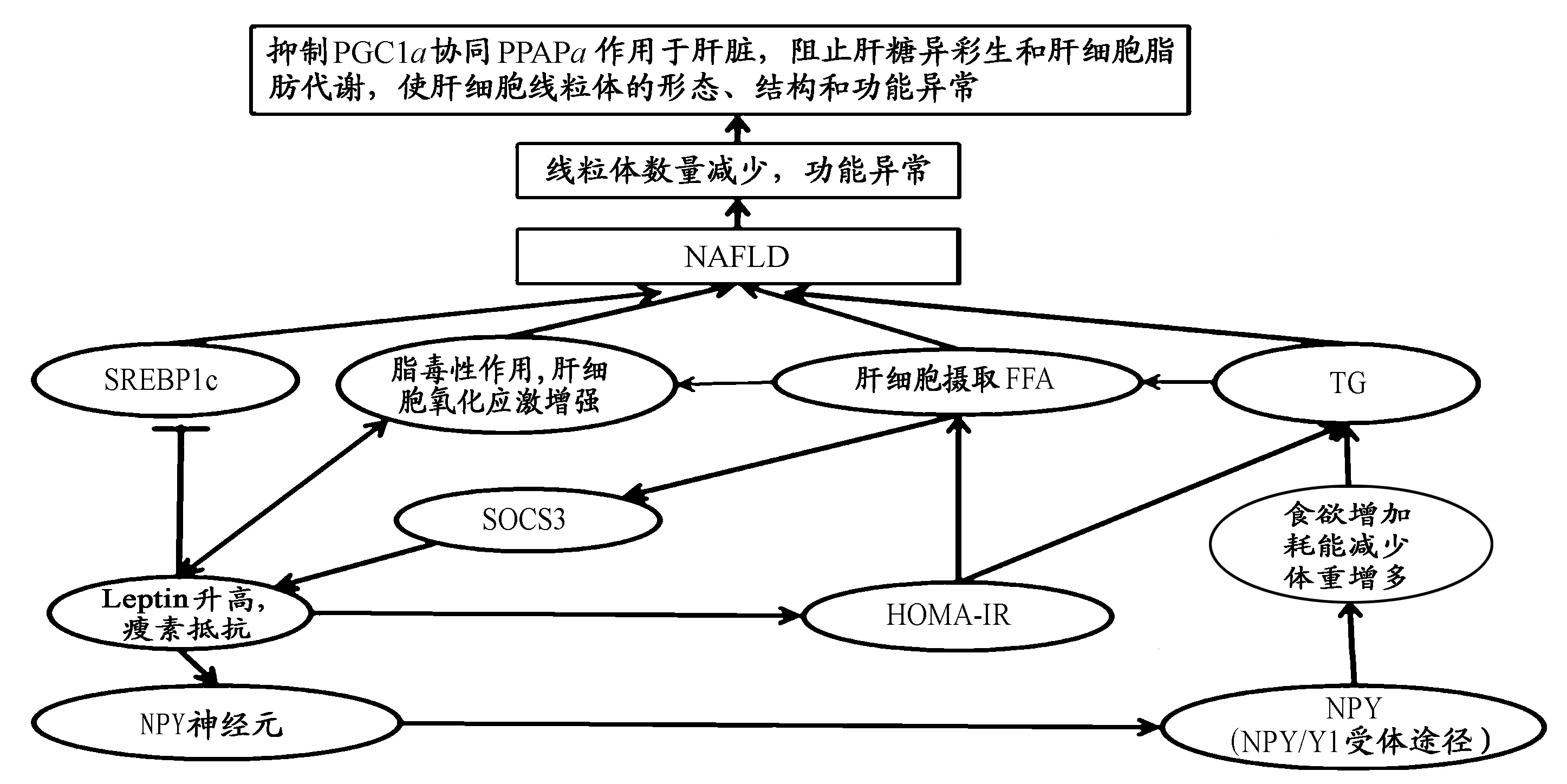
图 5 非酒精性脂肪肝大鼠肝组织脂肪变性的可能机制示意图
SREBPs是一类位于内质网上的膜连接蛋白,SREBP1c是参与脂肪合成基因的主要调节因子,对正常非脂肪组织脂肪含量的稳定起着至关重要的作用,此过程受Leptin的抑制[3],SREBP1c主要与脂肪酸代谢和糖代谢有关。若出现瘦素抵抗等瘦素作用缺乏的情况,而胰岛素含量增多或作用增加,SREBP1c将会出现过表达,脂肪合成相关的酶水平或活性增加,TG含量增多,造成脂肪在非脂肪组织(如肝脏)的异常蓄积,形成脂肪肝。在肝脏、脂肪组织中SREBP1c/ SREBP1a的比值是9.3,可见,SREBP1c是肝组织固醇调节元件结合蛋白的主要亚型[3]。SREBP1c是肝脏脂质代谢的关键调控因子,参与几乎所有的肝脏TG和脂肪酸合成基因的转录。有研究表明[16],在发生NAFLD时,SREBP1c表达增强,几乎是对照组的5倍,而SREBP1c转基因鼠则可自然发生NAFLD。Kakuma等[15]实验证明,fa/fa肥胖大鼠肝脏、胰腺的SREBP1c表达增加,而用基因沉默技术造成瘦素抵抗的同种大鼠肝脏SREBP1c基因表达显著降低。在本实验中,高脂对照组大鼠与正常对照组相比,SREBP1c基因表达量增加8.68倍,与本研究结果类似的研究有Tobe等[23]对IRS2-/-的IR小鼠研究,发现肝脏SREBP1c基因转录明显增加。这一情况在实验初期血糖水平正常的病程早期也不例外,表明,SREBP1c基因表达增加既不是由高血糖引起,也不是由高胰岛素引起。Tobe等[23]的研究中用高瘦素处理小鼠后,小鼠出现了摄食减少和体重减轻,同时SREBP1c基因的过度表达也得到了较好的控制,这一实验结果与本研究相似。在本实验的NAFLD大鼠模型中存在一定程度的瘦素抵抗,且由于瘦素抵抗抑制了SREBP1c在肝脏的正常脂质代谢功能,进一步引起肝脏这一非脂肪组织脂肪累积,形成一个恶性循环。FE组大鼠的SREBP1c基因表达出现了明显的降低,提示可能是通过运动刺激使得大鼠恢复对瘦素的敏感性,进而恢复瘦素对SREBP1c的负调控。
SOCS3升高是瘦素抵抗的标志[4],Lagathu等[18]研究发现,增多的FFA可通过使SOCS3高表达加重IR。Ueki等[24]在实验中发现,SOCS3参与NAFLD的发生,不仅是由于SOCS3作用于胰岛素信号通路,从而导致脂质代谢紊乱,SOCS3还可上调SREBP1c的表达,使肝脏这一非脂肪组织的脂肪蓄积异常。在本实验(图5)和此前的研究中发现[1],瘦素抵抗可能是NAFLD独立的发病危险因素,这一点在实验中依然存在,SOCS3的高表达标志着瘦素抵抗的存在,FC组SOCS3 mRNA基因的表达是C组的6倍多,FC组存在明显的瘦素水平升高。这说明,NAFLD进程中存在的高瘦素水平,且SOCS3水平的增加也从另一面证实了其中存在的瘦素抵抗。SOCS3的高表达可以使STAT下调,同时STAT是抑制SREBP1c启动活性的分子,SOCS3水平的升高促使SREBP1c mRNA表达量增加,从而启动一系列脂肪酶的活性,从而使脂肪酸合成增多。而FE组大鼠通过运动刺激明显减轻了瘦素抵抗现象,使得肝脏超微结构中线粒体的数量和功能有了较明显地恢复。
过氧化物酶体增殖剂激活受体(peroxisome proliferators-activated receptors,PPARs)属于核受体家族中配体激活的转录因子,有3种亚型,与脂质代谢、葡萄糖代谢、脂蛋白代谢、细胞增殖等关系密切。PPARα主要在肝脏中表达,参与调节脂肪酸氧化的几种关键酶的表达,通过诱导线粒体和过氧化物酶体氧化水平及相关基因表达来刺激脂肪代谢,保护肝脏,避免肝细胞脂肪变性。PGC1α属于转录辅助活化因子家族,也是脂肪细胞分化的重要调节因子,PGC1α在肝脏中的主要作用是促进肝糖异生、调节肝细胞脂肪代谢。PPARα与PGC1α的共同作用是PGC1α调节肝细胞脂肪酸氧化的重要途径。在饥饿、寒冷或禁食状态下,PGC1α能激活肝脏脂肪酸氧化中PPARα目标基因的表达,PGC1α基因沉默小鼠肝脏脂肪酸氧化率降低,引起肝细胞脂肪变性,PGC1α-/-小鼠参与氧化的PPARα基因没有下调,而肝细胞线粒体功能下降。这提示,可能是由于线粒体功能下调而引起PGC1α-/-小鼠出现脂肪酸氧化率降低。PGC1α可与SREBP竞争HNF4α结合位点,干扰PGC1α的募集,从而抑制糖异生[28]。PGC1α是PPAR的辅助激活因子,协同PPARα作用于肝脏的脂质代谢、与脂质运动功能相关蛋白的调节,在生理状态下保护肝脏,防止肝细胞出现脂肪变性。本实验显示,FC组大鼠均出现了PPARα mRNA基因表达的上升,可能是由于肝脏中肝细胞的脂肪变性引起了PPARα mRNA基因表达的升高,以求肝脏能够尽可能的将多余的脂肪酸氧化利用,同时伴随出现了PGC1α mRNA基因表达水平的下调。由前面的肝脏超微结构观察可得出肝细胞脂肪变性,引起了肝细胞内线粒体功能障碍,线粒体数目和活性均受到影响,进而引起PGC1αmRNA基因表达的下调。由于PGC1α mRNA基因表达的下调在一定程度上也影响了PPARα的表达,引起肝细胞脂肪酸氧化障碍,加重NAFLD中存在的肝细胞脂肪变性。而运动在很大程度上使PGC1α得以恢复,PGC1α协同PPARα作用于肝脏,促进肝糖异生、肝细胞脂肪代谢,使肝细胞线粒体的形态、结构和功能趋于正常。
4 结论
1.16周高脂饮食NAFLD大鼠肝脏出现了明显的肝脏脂质异常蓄积,肝组织结构紊乱,肝细胞内线粒体数量减少、结构畸形。
2.规律的、强度适宜的有氧运动可明显改善NAFLD大鼠中存在的脂质代谢紊乱,可能通过促进肝脏细胞内线粒体的数量、形态结构、功能趋于正常,从而减少肝脏这一非脂肪组织的脂肪蓄积,减轻肝脏脂肪变性程度,促进非酒精性脂肪肝的良性转归。
[1]刘倩倩,肖国强.游泳运动对肥胖及肥胖抵抗型NAFLD大鼠干预效果的对比研究[J].体育学刊,2013,20(1):129-134.
[2]秦智.亚低温状态下游泳运动预防与改善SD大鼠NAFLD效果及机制实验研究[D].广州:华南师范大学,2010.
[3]童国玉,李果.固醇调节元件结合蛋白1c的研究进展[J].国外医学内分泌学分册,2002,22(5):328-331.
[4]中华医学会肝脏病学分会脂肪肝和酒精性肝病学组.非酒精性脂肪肝病诊断标准[J].中华肝脏病学杂志,2003,11(2):71.
[5]ADAMS L A,ANGULO P.Treatment of nonalcoholic fatty liver disease[J].Postgrad Med J,2006,82(967):315-322.
[6]AL-HADDAD M,KHASHAB M,ZYROMSKI N,etal.Risk factors for hyperechogenic pancreas on endoscopic ultrasound:A case-control study[J].Pancreas,2009,38(6):672-675.
[7]BERGE R K,BJØMDAL B,STRAND E,etal.Tetradecylthiopropionic acid induces hepatic mitochondrial dysfunction and steatosis,accompanied by increased plasma homocysteine in mice[J].Lipids Health Dis,2016,15(1):1-16.
[8]CHOI C W,KIM G H,KANG D H,etal.Associated factors for a hyperechogenic pancreas on endoscopic ultrasound[J].World J Gastroenterol,2010,16(34):4329-4334.
[9]CICCONE M M,CORTESE F,GESUALDO M,etal.Dietary intake of carotenoids and their antioxidant and anti-inflammatory effects in cardiovascular care[J].Mediat Inflamm,2013:782137.DOI:10.1155/2013/78213.
[10]COHEN J C,HORTON J D,HOBBS H H.Human fatty liver disease:Old questions and new insights[J].Sci,2011,332(6037),1519-1523.
[11]DAY C P,JAMES O F.Steatohepatitis:A tale of two “hits”?[J].Gastroenterology,1998,114(4):842-845.
[12]GAGGINI M,MORELLI M,BUZZIGOLI E,etal.Non-alcoholic fatty liver disease (NAFLD) and its connection with insulin resistance,dyslipidemia,atherosclerosis and coronary heart disease[J].Nutrients,2013,5(5):1544-1560.
[13]GTATTAQLIANO L,DE BARI O,BERNARDO T C,etal.Role of mitochondria in nonalcoholic fatty liver disease-from origin to propagation[J].Clin Biochem,2012,45(9):610-618.
[14]HAGA Y,KANDA T,SASAKI R,etal.Nonalcoholic fatty liv-er disease and hepatic cirrhosis:Comparison with viral hepatitis-associated steatosis[J].World J Gastroenterol,2015,21(46):12989-12995.
[15]KAKUMA T,LEE Y,HIGA M,etal.Leptin,troglitazone,and the expressing of sterol regulatory element binding proteins in liver and pancreatic islets[J].Proc Natl Acad Sci USA,2000,97(15):8536-8541.
[16]KOHJIMA M,HIGUCHI N,KATO M,etal.SREBP-1c,regulated by the insulin and AMPK signaling pathways,plays a role in nonalcoholic fatty liver disease[J].Int J Mol Med,2008,21(4):507-511.
[17]KOLIAKI C,RODEN M.Hepatic energy metabolism in human diabetes mellitus,obesity and non-alcoholic fatty liver disease[J].Mol Cell Endocrinol,2013,379(1-2):35-42.
[18]LAGATHU F,BASTARD J P,AUCLAIR M,etal.Chronic interleukin-6 treatment increased IL-6 secretion and induced insulin resistance in adipocyte:Prevention by rosiglitazone[J].Biochem Biophys Res Commun,2003,311(2):372-379.
[19]MISRA A,JAISWAL A,SHAKTI D,etal.Novel phenotypic markers and screening score for the metabolic syndrome in adult Asian Indians[J].Diabetes Rese Clini Pract,2008,79(2):1-5.
[20]RHEE E J,LEE W Y,CHO Y K,etal.Hyperinsuliemia and the development of nonalcoholic Fatty liver disease in nondiabetic adults[J].Am J Med,2011,124(1):69-76.
[21]TILG H,MOSCHEN A R.Evolution of inflammation in non-alcoholic fatty liver disease:The multiple parallel hits hypothesis[J].Hepatology,2010,52(5):1836-1846.
[22]TOBE K,OGURA T,TSUKAMOTO C,etal.Relationship between serum leptin and fatty liver in Japanese male adolescent university students [J].AM J Gastroenterol,1999,94(11):3328-3335.
[23]TOBE K,SUZUKI R,AOYAMA M,etal.Increased expression of the sterol regulatory element binding protein binding protein(SREBP)-1c gene in insulin receptor substrate-2(-/-)mouse liver[J].J Biol Chem,2001,276(42):38337-38340.
[24]UEKI K,KONDO T,TSENG Y H,etal.Centrol role of suppressor of cytokine signaling proteins in hepatic steatosis,insulin resistance,and the metabolic syndrome in the mouse[J].Proc NatI Acad Sci U S A,2004,101(28):10422-10427.
[25]WANG Z,ZHOU Y T,KAKUMA T,etal.Leptin resistance of adipocytes in obesity:Role of suppressors of cytokine signaling[J].Biochem Biophys Res Commun,2000,277(1):20-26.
[26]WANLESS I R,SHIOTA K.The pathogenesis of nonalcoholic steatohepatitis and other fatty liver diseases:A four-step model including the role of lipid release and hepatic venular obstruction in the progression to cirrhosis[J].Semin Liver Dis,2004,24(1):99-106.
[27]WILLIAMSON R M,PRICE J F,GLANCY S,etal.Prevalence of and risk factors for hepatic steatosis and nonalcoholic Fatty liver disease in people with type 2 diabetes:The Edinburgh Type 2 Diabetes Study[J].Diabetes Care,2011,34(5):1139-1144.
[28]YAMAMOTO T,SHIMANO H,NAKAGAWA Y,etal.SR-EBP-1 interacts with HNF-4a and interferes with PGC-1 recruitment to suppress hepatic gluconeogenic genes[J].J Biol Chem,2004,279(13):12027-12035.
[29]YANG S,ZHU H,LI H,etal.Mitochondrial adaptations to obesity2 related oxidant stress[J].Arch Biochem Biophys,2000,378(2):259-268.
Impact of Aerobic Exercise on the Function and Mitochondrial Structure of Liver Cell in NAFLD Rats
LIU Qian-qian1,QIN Zhi2,HOU Gai-xia1,XI Xue-feng1,XIAO Guo-qiang3
Objective:To observe the effects of eight weeks of swim-training on the abnormal accumulation of liver lipid,disorders of function and mitochondrial structure in NAFLD rats.Methods:Dividing 35 rats (3 month old) SPF male SD rats into normal control group (C group,9 rats,fed with standard diet) and high fat diet group (26 rats at random,fed with high fat diet to build the NAFLD model).Dividing the high fat diet group into high fat control group (FC,13 rats) and high fat exercise-training group (FE,13 rats) at random.The exercise training groups had an 8 weeks swimming training (6 days in one week).After the training,measuring liver index and serum lipid levels,using microscope to measure liver steatosis,observing the structure and number changes of liver cell mitochondria by electron microscope.Detecting the mRNA levels of SOCS3,SREBP1c,PPARα,PGC-1α by ORT-PCR.Results:Compared with the C group,body weight,liver weight,liver fat,TC,FFA,SREBP1c,the expressions of SOCS3 mRNA in FC group significantly increased (P<0.01 orP<0.05),HDL-c reduced (P<0.05);compared with the FC group,body weight,liver weight,liver index,TC,FFA,SREBP1c,and the expressions of SOCS3 mRNA in FE group significantly decreased (P<0.01 orP<0.05).The microscopic structure showed that in C group,hepatic lobular had clear structure,the hepatic cord arranged orderly,the liver cells had no fat vacuoles,mitochondrial cristae was rich and well-organized;in FC group,hepatic lobule structure disappeared,with nucleus abnormal,perinuclear edema,and mitochondria swelling,cristae was disappear or arranged chaos,the number of intracellular lipid droplets and changed vacuolar significantly increased;in FE group,hepatic lobular and hepatic cord had clear structure,membrane structure was normal,mitochondria and endoplasmic reticulum were relatively dense,intracellular lipid droplets and vacuolar degeneration appeared,mitochondrial cristae structure was relatively obvious.Conclusion:1) after 16 weeks’ high fat diet,the NAFLD rats’ liver had obviously abnormal accumulation of liver lipid,with disorder of liver structure,the number of mitochondria in liver cells decreased,with mitochondria’s structural abnormalities;2) Regular and appropriate intensity aerobic exercise can significantly improve lipid metabolic disorder in NAFLD rats,which probably promoting mitochondria number,morphology,structure and function of liver cell to be normal,then reducing fat accumulation in the liver,relieving liver fatty degeneration,and promoting the benign outcome of non alcoholic fatty liver.Key words:NAFLD;fat-freemass;livercell;mitochondrialstructure;adiposedegeneration;rat
1002-9826(2016)05-0075-08
10.16470/j.csst.201605011
2015-04-28;
2016-07-05
河南省教育厅人文社会科学研究项目(2016-QN-178)。
刘倩倩(1986-),女,山东聊城人,讲师,博士,主要研究方向为慢性病的运动防治、运动负荷的生物学适应, E-mail:wwwstrom@sina.com。
1.河南大学 体育学院,河南 开封 475001;2.武汉体育学院,湖北 武汉 430079;3.华南师范大学,广东 广州 510006 1.Henan University,Kaifeng 475001,China.2.Wuhan Sports University,Wuhan 430079,China.3.South China Normal University,Guangzhou 510006,China.
G804.2
A
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31
传染病信息(2022年2期)2022-07-15
中国药学药品知识仓库(2022年9期)2022-05-23
中国典型病例大全(2022年13期)2022-05-10
昆明医科大学学报(2022年2期)2022-03-29
昆明医科大学学报(2022年1期)2022-02-28
中国现代医生(2016年30期)2017-03-02
中国当代医药(2016年33期)2017-02-27
医学信息(2016年36期)2017-02-23
中国实用医药(2016年8期)2016-03-30
