
水产品中喹烯酮和喹赛多及其主要代谢物残留的HPLC-MS/MS检测方法研究
2017-01-06 04:29孙建华孙振中刘菁华黄雪玲刘云璐
分析测试学报 2016年12期
郭 霞,孙建华,孙振中,刘菁华,黄雪玲,刘云璐,陈 琳
(上海市水产研究所,上海 200433)
水产品中喹烯酮和喹赛多及其主要代谢物残留的HPLC-MS/MS检测方法研究
郭 霞*,孙建华,孙振中,刘菁华,黄雪玲,刘云璐,陈 琳
(上海市水产研究所,上海 200433)

水产品;喹烯酮;喹赛多;代谢物;高效液相色谱-串联质谱(HPLC-MS/MS)

1 实验部分
1.1 仪器与试剂
TSQ QUANTUM ACCESS高效液相色谱-串联质谱仪(美国Thermo公司);电子天平(XS105,梅特勒-托利多仪器有限公司);高速冷冻离心机(Centrifuge 5810R,艾本德国际贸易有限公司);旋转蒸发仪(瑞士Buchi公司);Oasis MAX固相萃取柱(60 mg,3 mL,美国Waters公司);Milli-Q超纯水仪(法国Millipore公司)。
1.2 溶液的配制
标准溶液:准确称取适量的QCT,CYA,BDQCT,MQCA,BDCYA,QCA标准品,先用2 mL二甲亚砜溶解,再用甲醇稀释并定容,CYA先用2 mL N,N-二甲基甲酰胺溶解,再用甲醇稀释并定容,分别配制成质量浓度为100 μg/mL的标准物质储备溶液,-20 ℃保存,有效期1个月;混合标准工作溶液:准确移取适量6种分析物标准储备溶液,用甲醇稀释至质量浓度为10 μg/mL,-20 ℃保存,有效期1个月。
0.3 mol/L盐酸溶液:移取2.7 mL浓盐酸,加水稀释定容至100 mL;
0.2 mol/L磷酸盐缓冲液(pH 7.6):称取2.7 g磷酸二氢钾加700 mL水溶解,用0.05 mol/L氢氧化钾调至pH 7.6,加水稀释至1 L。

表1 梯度洗脱条件
1.3 色谱条件
色谱柱:Waters XBridge C18色谱柱(100 mm×2.1 mm i.d.,粒径3.5 μm);流速:0.25 mL/min;进样量:25 μL;流动相由乙腈、甲醇和0.1%甲酸溶液组成,梯度洗脱程序见表1。
1.4 质谱条件
离子源:电喷雾离子源(ESI);离子化方式:正离子;扫描方式:选择反应监测(SRM);电喷雾电压:4 000 V;离子传输管温度:350 ℃;鞘气流量:30 mL/min;辅助气流量:8 mL/min;碰撞气:氩气;其它优化的质谱条件见表2。

表2 喹烯酮和喹赛多及其主要代谢物的优化质谱条件
*quantitation ion
1.5 样品前处理
称取均质后的组织样品5.00 g(精确至0.02 g)于50 mL聚丙烯离心管中,加入15 mL乙腈-乙酸乙酯(1∶1)进行提取,涡旋混匀,振荡提取2 min,然后于4 ℃,6 000 r/min离心10 min,将上清液转移至100 mL鸡心瓶。向残渣中加入15 mL乙腈-乙酸乙酯(1∶1)提取液,重复提取1次,上清液转至同一鸡心瓶。残渣中加入10 mL 0.3 mol/L盐酸溶液,涡旋混匀2 min,超声提取10 min,然后于4 ℃,6 000 r/min离心10 min,上清液转移至另一离心管中,向离心管中加入15 mL乙酸乙酯振荡提取5 min,6 000 r/min离心10 min,上清液合并至鸡心瓶中,继续向离心管中加入15 mL乙酸乙酯重复提取1次,上清液转至鸡心瓶。向鸡心瓶中加入3 mL 0.2 mol/L的磷酸盐缓冲液(pH 7.6)后,于40 ℃旋转蒸发至约3 mL。
将MAX固相萃取柱依次用3 mL甲醇、3 mL 0.2 mol/L的磷酸盐缓冲液活化、平衡,将鸡心瓶中的溶液上样,自然重力下流出,向鸡心瓶中加入1 mL 0.2 mol/L的磷酸盐缓冲液清洗鸡心瓶,将清洗液过柱。3 mL水洗涤杂质后用4 mL 2%甲酸甲醇溶液洗脱,收集洗脱液,40 ℃下氮吹至干,加入1 mL 0.1%甲酸-乙腈(9∶1)溶液,涡动混匀,装入棕色进样小瓶,供HPLC-MS/MS测定。
1.6 标准曲线的绘制
用混合标准工作溶液和各组织样品经前处理后获得的基质空白溶液配制成2,5,10,50,100,500 μg/L的系列标准溶液,取25 μL进样分析。分别以各分析物的峰面积(Y)为纵坐标,质量浓度(X,μg/L)为横坐标绘制标准曲线,求回归方程及相关系数。各组织样品中分析物的浓度采用组织样品的基质添加标准曲线法测定。
2 结果与讨论
2.1 标准储备溶液的配制
在配制标准储备液过程中,向标准物质中先加入2 mL二甲亚砜进行溶解,再加入甲醇稀释并定容,发现QCT,BDQCT,MQCA,BDCYA和QCA的溶解度较好,得到均一透明的储备液。但CYA的溶解度较差,配制的储备液浑浊,影响了储备液的准确度和仪器检测灵敏度。后改用2 mL N,N-二甲基甲酰胺溶解CYA,再用甲醇稀释定容的方法可获得均一稳定的CYA标准储备液。
2.2 提取条件的选择
2.3 固相萃取柱的选择
残留检测的常用净化方法有液液萃取法和固相萃取柱法。液液萃取法的试剂用量相对较大,而固相萃取柱法采用的试剂量少,净化效果好,故本研究选用固相萃取柱法净化样品。文献常用的固相萃取柱有C18柱[18-19,21]、MAX柱[15,17,22-23]及HLB柱[9,11,20,24]等。本实验根据被分析物的性质选用MAX固相萃取柱净化样品,分别对上样液和洗脱液的种类及用量进行筛选和优化。MAX固相萃取柱是混合阴离子交换柱,是反相和强阴离子交换的复合模式,由于QCT,BDQCT,CYA和BDCYA的脂溶性较大,均利用反相机制在小柱上保留、洗脱,而MQCA和QCA利用离子交换机制被保留和洗脱。上样液选用0.2 mol/L磷酸盐缓冲液(pH 7.6),此时MQCA和QCA在碱性磷酸盐缓冲溶液中呈离子态,利用离子交换机制被保留在萃取柱上。MQCA和QCA在酸性溶液中呈分子态,能够从离子交换柱上被洗脱下来,实验选用不同体积的含2%甲酸的甲醇溶液和含2%甲酸的乙腈溶液作洗脱液进行比较,结果显示以4 mL 2%的甲酸甲醇作洗脱液得到的回收率最高,回收率均大于90%。
2.4 色谱条件的选择
2.5 质谱分析
采用流动注射方式单标进样,流速10 μL/min,在质荷比(m/z)为100~500范围内对6种待测物(5 μg/mL)进行一级质谱图扫描,确定QCT,BDQCT,MQCA,CYA,BDCYA及QCA的分子离子分别为m/z307.0,275.0,189.0,271.8,240.0和174.9。然后对其进行二级质谱分析,分别选取丰度最强的离子作为定量离子,以次强离子作为定性离子(见表2)。优化碰撞电压等其他质谱分析条件。图1为草鱼空白样品添加QCT,BDQCT,MQCA,CYA,BDCYA及QCA的特征离子色谱图,添加浓度为5 μg/kg。由图可见,样品中被分析物的保留时间处无杂峰干扰。

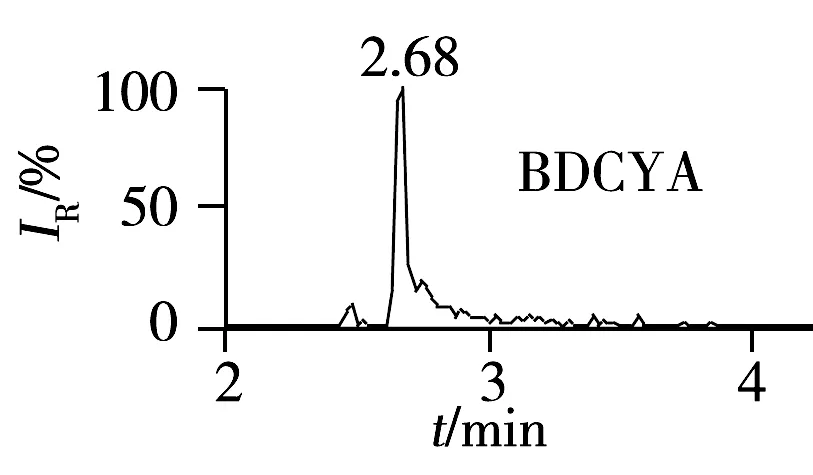
2.6 线性关系、检出限与定量下限
按照“1.6”绘制标准曲线,草鱼、南美白对虾、中华绒鳌蟹不同组织样品中6种分析物的基质添加标准曲线方程及相关系数见表3。从表3可见,6种目标分析物的浓度在2~500 μg/L范围内与其峰面积呈线性相关,相关系数均大于0.98。实验采用空白组织样品中添加目标组分的方法,按前处理步骤和仪器条件进行处理和检测,以特征离子色谱峰信噪比(S/N)大于3为检出限(LOD),大于10为定量下限(LOQ),确定不同组织中各化合物的检出限和定量下限(见表3),得不同组织中各化合物的LOD为0.5~1.6 μg/kg,LOQ为2.0~5.0 μg/kg,检出限较低。
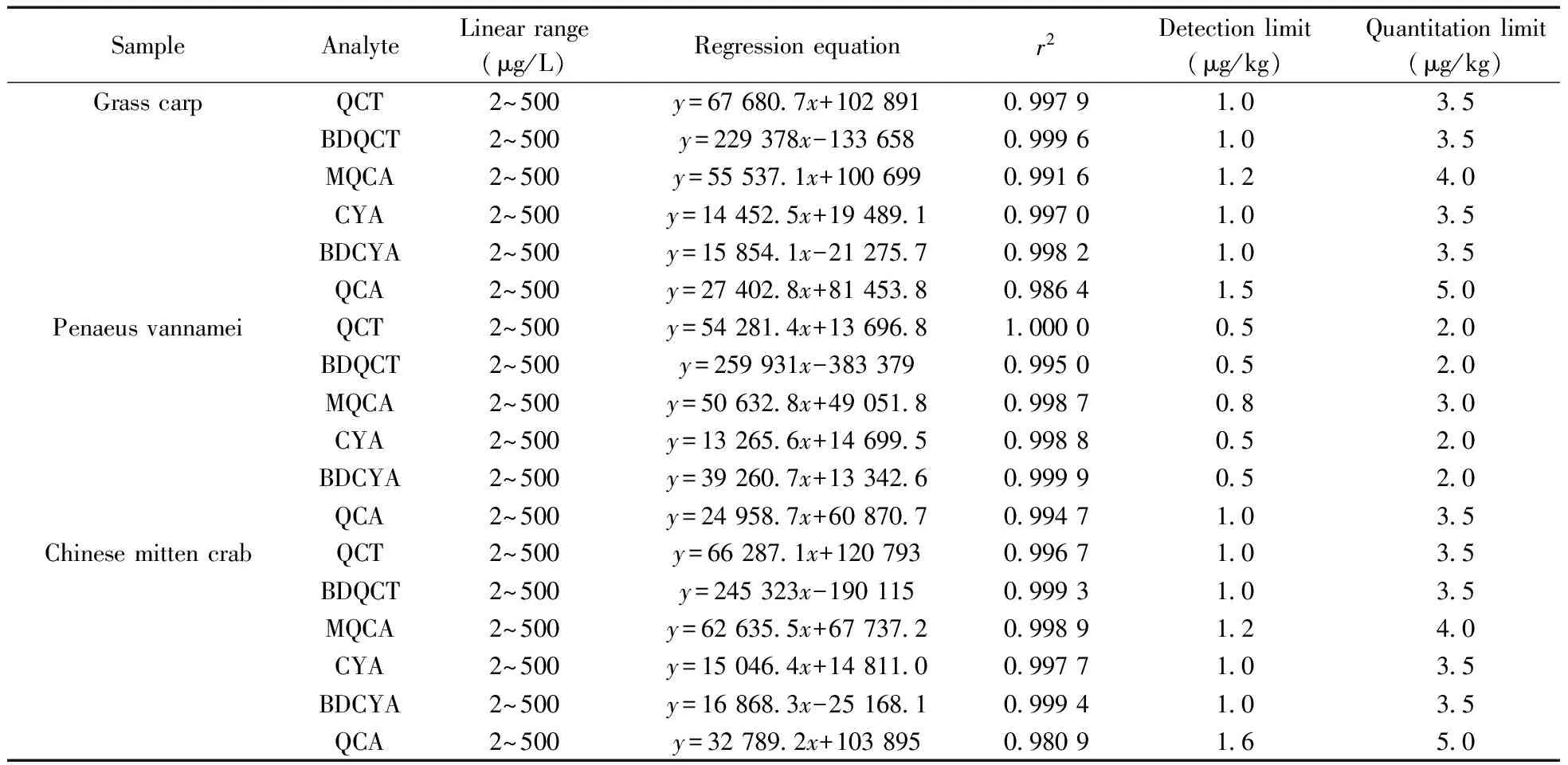
表3 不同组织中6种待测物的基质添加标准曲线、相关系数(r2)、检出限及定量下限
2.7 回收率与精密度
分别准确称取(5.00±0.02) g的草鱼、南美白对虾、中华绒鳌蟹空白样品组织,每种组织样品以5,10,50 μg/kg进行3个浓度的加标回收实验,将不同体积的混合标准工作液添加到空白样品中,静置15 min后开始提取。每个浓度水平设5个平行,随机取3日进行测定,计算平均回收率、日内相对标准偏差(RSD)和日间RSD。结果表明,QCT,BDQCT,MQCA,CYA,BDCYA和QCA在不同样品组织中3个加标水平下的平均回收率为76.3%~94.2%,日内RSD为4.2%~10.9%,日间RSD为7.2%~11.7%,表4列出了不同基质样品中6种待测物的回收率数据。该方法稳定可靠,可满足残留分析方法的要求。
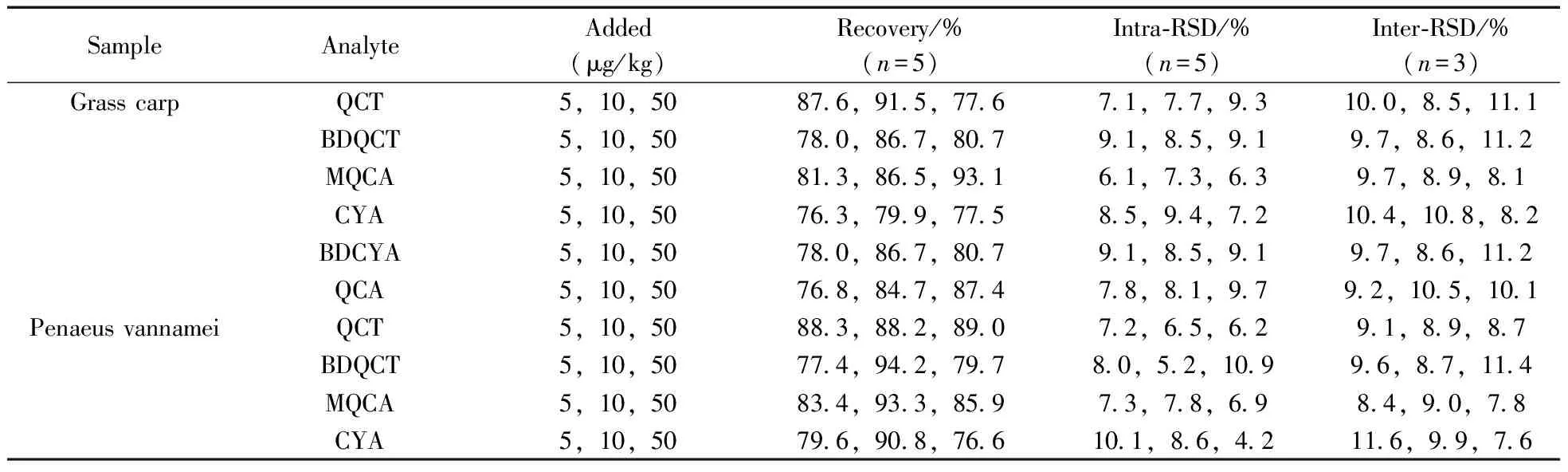
表4 不同组织中6种待测物的回收率及相对标准偏差
(续表4)

SampleAnalyteAdded(μg/kg)Recovery/%(n=5)Intra⁃RSD/%(n=5)Inter⁃RSD/%(n=3)BDCYA5,10,50791,880,76673,70,76104,84,81QCA5,10,50785,854,88359,70,8172,84,94ChinesemittencrabQCT5,10,50777,776,78681,72,91100,100,93BDQCT5,10,50781,841,79378,79,7993,94,115MQCA5,10,50831,845,85467,99,7086,117,77CYA5,10,50793,799,822105,94,83112,111,86BDCYA5,10,50893,917,86167,73,7290,100,85QCA5,10,50909,872,90248,51,5786,91,75
2.8 实际样品分析
3 结 论
本文采用HPLC-MS/MS法对水产品中残留的喹烯酮和喹赛多及其主要代谢物进行检测。方法采用乙酸乙酯-乙腈混合溶液、盐酸溶液分别提取样品中残留的目标物,经过MAX固相萃取柱净化,并对流动相的配比进行了优化,采用HPLC-MS/MS进行测定。在5~50 μg/kg加标水平内,方法的平均回收率为76.3%~94.2%,RSD为4.2%~11.7%,方法检出限为0.5~1.6 μg/kg,定量下限为2.0~5.0 μg/kg。本方法具有重复性好、准确稳定等特点,符合残留分析方法的性能要求,适于质检机构日常对水产品中喹烯酮和喹赛多及其主要代谢物残留的确证检测,而且本方法可同时检测2种原型药及其4种主要代谢物,与检测单一原型药物或单一代谢物的文献报道相比,可以更准确更全面地监测喹烯酮和喹赛多药物的残留。
[1] Zhang X Z,Liu Y,Xu Y J,Gong X H,Song X J,Liu H H,Tian X H,Liu Y H,Qin H W,Wang Z Q,Zhou Q L,Ren L H.J.FoodSaf.Qual.(张秀珍,刘云,徐英江,宫向红,宋向军,刘慧慧,田秀慧,刘义豪,秦华伟,王忠全,周全利,任利华.食品安全质量检测学报),2013,4(1):38-44.
[2] Fan L P,Duan Z J,Fang G Z,Tang Y W,Han H T,Wang S.J.Chin.Inst.FoodSci.Technol.(范立鹏,段振娟,方国臻,汤轶伟,韩海涛,王硕.中国食品学报),2012,12(9):171-178.
[4] Qiu Y M.TheResidueDepletionofCyadoxinSwine,ChickenandFish.Wuhan:Huazhong Agricultural University(邱玉敏.喹赛多在猪、鸡和鱼体内的残留消除研究.武汉:华中农业大学),2012.
[5] Hu G M.PharmacokineticsandResidueDepletionofQuinocetoneinCarp.Wuhan:Huazhong Agricultural University(胡桂敏.喹烯酮在鲤体内的药代动力学及残留研究.武汉:华中农业大学),2008.
[6] Yin J Y.PharmacokineticsandResidueDepletionofCyadoxinCarp.Wuhan:Huazhong Agricultural University(殷居易.喹赛多在鲤体内的药代动力学及残留研究.武汉:华中农业大学),2003.
[7] Wu S H,Zheng G M,Zhu X P,Chen K C,Dai X X,Pan D B.J.DalianFish.Univ.(吴仕辉,郑光明,朱新平,陈昆慈,戴晓欣,潘德博.大连水产学院学报),2009,24(S1):21-24.
[8] Yin Y,Li P J,Lin C,Liu S G,Zhu X P,Zheng G M,Chen K C,Liu Y H,Song Y,Wang Q.J.InstrumAnal.(尹怡,李平杰,林晨,刘书贵,朱新平,郑光明,陈昆慈,刘毅辉,宋怿,王群.分析测试学报),2013,32(11):1349-1353.
[9] Yang F,Liu Z C,Ye S S,Wu D F,Li Y P,Yu K J,Lin Y H,Qian J.Chin.J.Pharm.Anal.(杨方,刘正才,叶松生,吴德峰,李耀平,余孔捷,林永辉,钱疆.药物分析杂志),2008,28(4):630-633.
[10] Yang L,Ai X H,Yuan K P,Wang K Y.J.Instrum.Anal.(杨莉,艾晓辉,袁科平,汪开毓.分析测试学报),2010,29(4):372-375.
[11] Wang L,Ding H Z,Zhong J L,Jia H Q,Lu X X.Chin.AnimalHusbandryVeter.Med.(王林,丁焕中,钟家林,贾慧勤,鲁晓雄.中国畜牧兽医),2011,38(12):52-56.
[12] Qiu Y S,Yuan Z H,Fan S X,Liu D C,Huang L L,Yin J Y.Chin.J.Veter.(邱银生,袁宗辉,范盛先,刘登才,黄玲利,殷居易.中国兽医学报),2003,23(4):363-365.
[13] Xu Y J,Ren C B,Tian X H,Zou R J,Gong X H,Zhang X Z,Zhang H W,Yu Z Q,Liu Y M.J.Instrum.Anal.(徐英江,任传博,田秀慧,邹荣婕,宫向红,张秀珍,张华威,于召强,刘永明.分析测试学报),2011,30(10):1133-1137.
[14] Zhao S,Guo Q Z,Zhang J,Shao B.J.FoodSaf.Qual.(赵珊,郭巧珍,张晶,邵兵.食品安全质量检测学报),2013,4(1):124-128.
[15] GB/T 20746-2006.Method for Determination of the Residues of Carbadox,Olaquindox and Related Metabolites in Bovine and Porcine Liver and Muscle Tissues-LC-MS-MS Method.National Standard of the People's Republic of China(牛、猪肝脏和肌肉中卡巴氧、喹乙醇及代谢物残留的测定 液相色谱-串联质谱法.中华人民共和国国家标准).[16] Yin J Y,Ni M L,Shou C J,Fang K T,Xie D H,Li Z Q.Chin.J.Veter.Drug.(殷居易,倪梅林,寿成杰,房科腾,谢东华,李佐卿.中国兽药杂志),2006,40(1):11-15.
[17] Ouyang S,Pang G F,Xie L Q,Lan F,Lin L,Tu X K.J.Instrum.Anal.(欧阳姗,庞国芳,谢丽琪,蓝芳,林黎,涂小珂.分析测试学报),2008,27(6):590-594.
[18] Mei J L,Wu C M,Cheng L L,Shen J Z,Xue Q,Qian M Y,Shao M Y.Mar.Sci.(梅景良,吴聪明,程林丽,沈建忠,薛麒,钱民怡,邵梦瑜.海洋科学),2011,35(11):45-47.
[19] Lü H L,Wu C M,Cheng L L,Zhang S X,Shen J Z.Chin.J.Chromatogr.(吕海鸾,吴聪明,程林丽,张素霞,沈建忠.色谱),2012,30(1):45-50.
[20] Ye S S,Wu D F,Liu Z C,Yang F,Li Y P,Lin Y H,Yu K J,Lu S Y.J.FujianAgric.ForestryUniv.(叶松生,吴德峰,刘正才,杨方,李耀平,林永辉,余孔捷,卢声宇.福建农林大学学报),2008,37(3):307-311.
[21] Yi X B,Qiu L Q,Liu S Q,Huang X Q.J.Instrum.Anal.(易锡斌,裘立群,刘世琦,黄晓琴.分析测试学报),2015,34(3):346-351.
[22] Lin L,Xie Y Q,Ouyang S,Liang H,Ye G,Liao J J,Pang G F.Chin.J.Anal.Lab.(林黎,谢丽琪,欧阳姗,梁宏,叶刚,廖菁菁,庞国芳.分析试验室),2010,29(2):38-41.
[23] Xiao J Z,Ben Z,Hong Z,Xue C C,Guang M M.J.Sep.Sci.,2011,34:469-474.
[24] Zheng L,Wu Y J,Zhao Y F,Guo W,Chen Z G.J.Instrum.Anal.(郑玲,吴玉杰,赵永锋,郭蔚,陈智刚.分析测试学报),2014,33(1):21-26.
Research on the Residual Detection of Quinocetone,Cyadox and Their Main Metabolites in Aquatic Products by HPLC-MS/MS
GUO Xia*,SUN Jian-hua,SUN Zhen-zhong,LIU Jing-hua,HUANG Xue-ling,LIU Yun-lu,CHEN Lin
(Shanghai Fisheries Research Institute,Shanghai 200433,China)
A high performance liquid chromatography-tandem mass spectrometric(HPLC-MS/MS) method was established for the determination of the residual quinocetone(QCT),cyadox(CYA),bidesoxyquinocetone(BDQCT),methyl-3-quinoxaline-2-carboxylic acid(MQCA),bidesoxycyadox(BDCYA) and quinoxaline-2-carboxylic acid(QCA) in aquatic products.Samples were extracted with acetonitrile-ethyl acetate(1∶1) and hydrochloric acid solution,then cleaned up on a MAX solid-phase extraction cartridge.The separation of QCA,BDQCT,MQCA,CYA,BDCYA and QCT was carried out on a Waters XBridge C18column using methanol,acetonitrile and 0.1% fomic acid as mobile phase.The identification of six analytes was performed by selective reaction monitoring in positive electrospray ionization,and the quantification was done by the external standard method.The calibration curves of six analytes were linear in the range of 2-500 μg/L.At the fortified levels of 5-50 μg/kg,the recoveries ranged from 76.3% to 94.2%,with RSDs of 4.2% to 11.7%.The limits of detection(LOD) were in the range of 0.5-1.6 μg/kg and the limit of quantitation(LOQ) were 2.0-5.0 μg/kg.The results showed that the method could be used to analyze simultaneously QCT,CYA and their metabolites residues in aquatic products.
aquatic products;quinocetone(QCT);cyadox(CYA);metabolites;high performance liquid chromatography-tandem mass spectrometry(HPLC-MS/MS)
2016-03-10;
2016-07-10
上海市青年人才成长计划(沪农青字【2015】第3-10号)
10.3969/j.issn.1004-4957.2016.12.004
O656.63;S816.7
A
1004-4957(2016)12-1535-07
*通讯作者:郭 霞,硕士,工程师,研究方向:理化分析,Tel:021-65483215-669,E-mail:guoxia_great2013@126.com
猜你喜欢
食品安全导刊·中旬刊(2022年3期)2022-04-15
食品工程(2020年4期)2021-01-20
河北果树(2020年4期)2020-11-26
中国油脂(2020年3期)2020-04-10
小天使·三年级语数英综合(2019年9期)2019-11-09
创新作文(1-2年级)(2019年4期)2019-10-15
无机化学学报(2016年8期)2016-12-06
化学分析计量(2016年1期)2016-03-14
中国粮油学报(2016年1期)2016-02-06
化工进展(2015年3期)2015-11-11
