
分子印迹电化学传感器的制备及其快速检测饮水中草甘膦残留的应用研究
2017-01-06 04:29李腾飞赵风年王珊珊佘永新金茂俊刘海金郑鹭飞
分析测试学报 2016年12期
张 超,李腾飞,赵风年,王珊珊,佘永新*,金茂俊,刘海金,金 芬,邵 华,郑鹭飞,徐 平,王 静*
(1.中国农业科学院 农业质量标准与检测技术研究所,北京 100081;2.西藏自治区农畜产品质量安全检验检测中心,西藏 拉萨 850000)
分子印迹电化学传感器的制备及其快速检测饮水中草甘膦残留的应用研究
张 超1,李腾飞1,赵风年1,王珊珊1,佘永新1*,金茂俊1,刘海金2,金 芬1,邵 华1,郑鹭飞1,徐 平2,王 静1*
(1.中国农业科学院 农业质量标准与检测技术研究所,北京 100081;2.西藏自治区农畜产品质量安全检验检测中心,西藏 拉萨 850000)
以吡咯(Py)为功能单体,草甘膦(Gly)为模板,采用电化学聚合法构建了草甘膦分子印迹电化学传感器。通过循环伏安法(CV)、差分脉冲伏安法(DPV)、电化学交流阻抗法(EIS)对印迹电极性能进行了表征,筛选了印迹电极的聚合体系和模板分子的洗脱方法,优化了检测体系的pH值和吸附时间等。结果表明,以铁氰化钾为电活性探针,在最优检测体系中该印迹传感器对草甘膦具有特异性快速响应、灵敏度高和稳定性好的优点,传感器的峰电流与草甘膦浓度在5~800 ng/mL范围内呈良好的线性关系,相关系数(r2)为 0.981 7,检出限(S/N=3)为0.27 ng/mL。该传感器具有良好的重现性和稳定性,放置3周后对目标物的响应峰电流无明显变化。用于实际样品中草甘膦的测定,加标回收率为 78.6%~99.0%,能满足现场快速检测的要求。
吡咯;草甘膦;分子印迹电化学传感器;饮水
草甘膦(Glyphosate,Gly)是一种内吸传导型广谱灭生性除草剂,其作用机理主要是抑制植物体内的烯醇丙酮基莽草素磷酸合成酶,从而抑制莽草素向苯丙氨酸、酪氨酸及色氨酸的转化,使蛋白质合成受到干扰,导致植物死亡[1]。该类除草剂具有广谱高效、低毒、易分解等特性,广泛应用于果园、桑园、茶园、胶园等多种领域。目前,草甘膦是世界上应用最广、产量最大的农药品种,年销售量高居农药之首。2015年“草甘膦致癌风波”和2016年3月德国啤酒中被检出草甘膦(含量在0.49~29.74 μg/L之间)等事件[2],进一步助推了国际社会对草甘膦安全风险的极大关注。中国是草甘膦的第一大生产国和出口国,随着抗草甘膦植物的推广和抗草甘膦杂草的增加,其使用量逐年增加,对农作物、生态环境和人类健康可能带来一定的安全风险。因此,开展水环境中草甘膦残留的研究具有重要现实意义。
世界各国对饮水中草甘膦的最大残留量(MRL)均制定了限量规定,美国环境保护协会(US-EPA)制定饮水中草甘膦的MRL为700 μg/L[3];加拿大规定饮水中草甘膦最大可接受限量(MAC)为280 μg/L[4];德国规定饮用水中Gly极值不可超过0.1 μg/L[5];我国在《生活饮用水卫生标准》(GB5749-2006)规定了饮用水中Gly限量为700 μg/L[6]。目前,草甘膦的检测方法主要有高效液相色谱法[7]、高效液相色谱-串联质谱法[8]、气相色谱法、气相色谱-串联质谱法[9]、毛细管电泳法[10]、离子色谱法[11]、酶联免疫法[12]和电化学发光法[13]等。由于草甘膦极性较大,难溶于有机溶剂,缺乏可用于检测的官能团,采用LC和GC色谱方法检测均需衍生化;离子色谱法干扰因素较多,对草甘膦的检测结果会产生一定的影响;ELASA等快速检测技术存在稳定性差和灵敏度不高等缺点。因此,为满足饮水中草甘膦的痕量检测,研发操作简便、成本低、灵敏度高、特异性好的快速检测方法显得尤为重要。
分子印迹聚合物(Molecularly imprinted polymers,MIPs)具有形状、尺寸及功能基团与目标分子相匹配的印迹位点,对目标分子具有特异识别性能,因抗恶劣环境能力强、重复使用、稳定性好等优点,在传感器技术、环境检测、医药领域等方面展现出良好的应用前景[14-15]。MIPs作为特异性识别元件与电极相结合,可制备出对目标分子具有高度选择识别性的分子印迹电化学传感器,已被用于敌草隆[16]、环嗪酮[17]、绿麦隆[18]、速灭威[19]等多种农药残留的检测。分子印迹传感器通常存在着模板分子洗脱困难和聚合膜稳定性差的问题。吡咯具有良好的导电性、氧化还原性及局部交联的特性,作为功能单体可以通过掺杂和脱掺杂形成稳定性好、吸附性能强的疏松多孔结构的聚吡咯分子印迹聚合膜,该聚吡咯分子印迹聚合膜具有模板分子快速固载、洗脱和传质等优点。本文以吡咯为功能单体,草甘膦为模板分子,通过电化学聚合和聚吡咯过氧化法,在电极表面合成了聚吡咯多孔印迹膜,制备了草甘膦分子印迹电化学传感器,以铁氰化钾为活性探针,筛选和优化关键参数,构建了草甘膦残留检测方法学体系,实现了饮水中草甘膦残留的快速检测。
1 实验部分
1.1 仪器与试剂
CHI630E电化学工作站(上海辰华仪器公司);KQ2200DB3L超声波清洗仪(昆山市超声仪器有限公司);DF-101水浴锅(巩义市予华仪器有限公司);S-4800扫描隧道显微镜(日本Hitachi公司);Milli-Q超纯水系统(法国Millipore公司)。采用三电极体系,金电极(直径为3 mm)为工作电极,铂网电极为对电极,饱和甘汞电极为参比电极(天津艾达恒晟科技有限公司)。
草甘膦、氨甲基膦酸、吡咯(阿拉丁试剂有限公司);毒死蜱、涕灭威(Dr.Ehrenstoefer GmbH);5 mmol/L铁氰化钾溶液:K3[Fe(CN)6]0.164 5 g,K4[Fe(CN)6] 0.211 2 g,KCl 0.745 5 g 溶于100 mL 0.01 mol/L PBS溶液;Britton-Robison缓冲溶液(BR):0.04 mol/L磷酸、硼酸和醋酸配成,用0.2 mol/L的NaOH调节pH值。所有实验均在室温下进行。
1.2 分子印迹电极及非印迹电极的制备
将金电极依次用 0.3,0.05 μm Al2O3粉抛光,再依次用无水乙醇和二次蒸馏水超声清洗,每次5 min,用N2吹干。
采用循环伏安法在金电极表面制备分子印迹膜:聚合溶液含有13 μL吡咯(36.98 mol/L)、1 mL草甘膦(7.396 mol/L)和4 mL BR缓冲液(0.04 mol/L,pH 5.0),超声2 min,在-1.0~1.0 V 电位范围内,以100 mV·s-1速率循环扫描5圈。采用过氧化法去除模板分子:将所制电极置于0.1 mol/L NaOH溶液中,采用循环伏安法,在-1.3~1.2 V电位范围内,以100 mV·s-1速率循环扫描 20 圈,形成过氧化聚吡咯,从而洗脱模板分子。采用相同的制备条件,不加入草甘膦模板,制备非印迹膜电极。制备过程如图1所示。

图1 草甘膦分子印迹电极的制备及检测过程图Fig.1 Schematic representation of the preparation of imprinted electrode and the process of detection
1.3 实验方法
采用标准三电极检测体系:印迹传感器为工作电极,铂丝电极为辅助电极,饱和甘汞电极为参比电极。循环伏安和差分脉冲伏安法(DPV)表征电极均采用含有 5 mmol/L K3Fe(CN)6的 0.5 mol/L KCl溶液。循环伏安法表征参数:起始电压-0.2 V;终止电压+0.6 V;扫速50 mV·s-1;差示脉冲法表征参数:起始电压-0.2 V;终止电压+0.6 V;扫速50 mV·s-1。
将去除模板分子的印迹电极先在空白底液0.2% 甲酸水溶液(pH 4.0)中进行多次CV扫描(-0.2~0.6 V),直至曲线稳定。然后将电极浸入含有一定浓度Gly的1/1 000甲酸水溶液(pH 4.0)中,富集18 min后,取出电极,用超纯水反复冲洗电极表面,去除非特异性吸附到电极表面的物质,再将电极置入铁氰化钾溶液中进行电化学检测。
2 结果与讨论
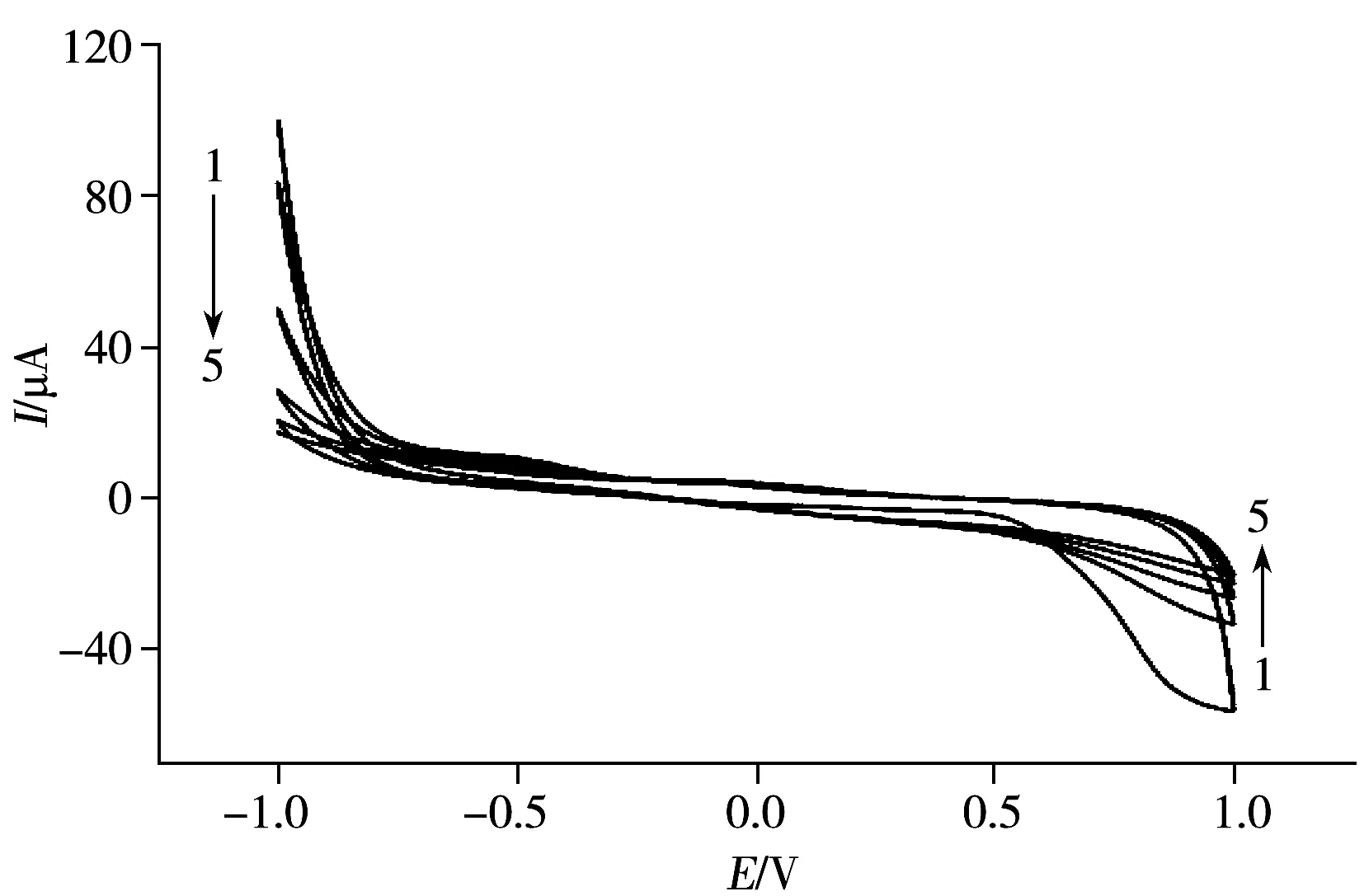
图2 草甘膦分子印迹膜电聚合过程的循环伏安曲线Fig.2 Cyclic voltammograms for electropolymerization of imprinted film in the presence of glyphosate
2.1 分子印迹电极制备体系的优化
2.1.1 聚合体系筛选 以吡咯作为功能单体时,聚合物体系是影响阴离子的目标物掺杂过程的重要因素。本实验研究了电极在BR缓冲液(pH 5.0)、0.2 mol/L硫酸和PBS缓冲液(pH 7.0) 3种聚合体系中的印迹效果。结果表明:工作电极在BR缓冲液(pH 5.0)聚合体系下制备的分子印迹膜对草甘膦具有很好的识别能力,此时草甘膦以负离子形式被成功掺杂到含正电聚吡咯网络中,而在其他缓冲液条件下合成的膜对草甘膦无响应。因此,本实验确定以BR缓冲液(pH 5.0)为最佳聚合体系。
2.1.2 聚合溶液模板与吡咯单体配比的选择 功能单体与模板分子的比例对印迹膜的识别性能和形貌有影响。通过改变草甘膦与吡咯的浓度比(1∶3,1∶5,1∶8)制备了不同的印迹聚合膜,比较了这些电极在相同条件下分别吸附同一浓度(100 ng/mL)草甘膦溶液的效果。结果显示,当功能单体与模板分子的比例为1∶5时,印迹电极对目标物的响应电流变化最大,此时印迹电极能够形成更多有效的识别位点。

图3 草甘膦分子印迹膜洗脱过程的循环伏安曲线Fig.3 Cyclic voltammograms for the elution of imprinted electrode

图4 不同电极在含 5 mmol/L K3Fe(CN)6的0.5 mol/L KCl溶液中的循环伏安图Fig.4 Cyclic voltammograms of different electrodes in solution containing 5 mmol/L K3Fe(CN)6and 0.5 mol/L KCla.bare gold electrode;b.MIP-modified gold electrode after elution;c.MIP-modified gold electrode/NIP-modified gold electrode
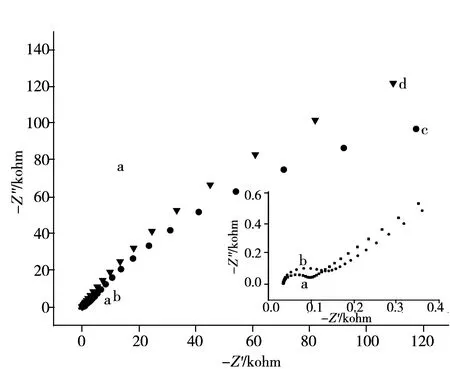
图5 不同草甘膦分子印迹电极的交流阻抗图Fig.5 Electrochemical impedance spectra of different electrodesa.bare gold elecrode,b.MIP-modified gold electrode after elution,c.NIP-modified gold electrode,d.MIP-modified gold electrode;insert:partial enlarged drawing of a and b
2.1.3 印迹电极的循环伏安特性及电化学行为表征 图2是含模板分子Gly的聚合液在金电极表面聚合过程中的CV曲线。由图可知,从第2圈开始,在电位为-0.3 V和+0.9 V处,金电极在聚合体系中的氧化还原峰电流均逐渐减小,随着扫描圈数的增加,金电极的氧化还原峰电流缓慢减小,直至曲线趋于平直,说明在电位-1.0~+1.0 V之间,聚吡咯膜由于阴离子模板分子的掺杂,在金电极表面逐渐由导电膜转变为钝化的不导电聚合膜,阻碍了溶液与电极之间的电子传递,从而使电流响应值降低。
当聚吡咯印迹电极在-1.3~1.2 V电位范围内,浸入 0.1 mol/L NaOH溶液中进行循环伏安扫描而被过氧化后,由于溶液中的强亲核试剂OH-与聚吡咯环上活性位点的相互作用,促使含氧基团羰基或羧基被引入聚吡咯结构单元,对阴离子模板分子产生排斥,实现了模板分子的脱掺杂[20]。图3是印迹电极洗脱过程的CV曲线。由图可见0.8~1.0 V 是PPy的氧化电位,随着扫描圈数的增加,-0.3 V与0.9 V 处的电流逐渐增大,说明聚吡咯膜在碱性条件下被过氧化后可促进Gly模板分子脱掺杂过程,形成了新的电子传递通道[21]。
以 5 mmol/L K3Fe(CN)6(含0.5 mol/L KCl)溶液为电化学信号探针,采用循环伏安法对不同电极进行表征,如图4所示。裸金电极(曲线a)有最大的氧化还原峰电流,印迹电极聚合曲线和非印迹电极聚合曲线(曲线c)重合,几乎无峰电流。而洗脱模板分子后的印迹电极(曲线b)峰电流介于曲线a与曲线c之间,说明洗脱模板分子后,形成的印迹空穴形成了电子传递通道,有利于探针在印迹电极表面发生氧化还原反应而产生信号。
本实验进一步采用电化学阻抗技术(EIS)对不同电极进行表征。在 EIS 图中,半圆的直径越大,表示其阻抗越大,相应的电子转移越难。如图5所示,裸金电极(曲线a)有很大的峰电流绝对值,其EIS半径最小,交流阻抗最小,这是因为裸金电极表面无阻止探针电子传递的聚合膜;非印迹电极(曲线c)与印迹电极(曲线d)由于表面均覆盖了钝化的聚合膜,电子探针转移受阻,所以EIS半径均比裸金电极大。而印迹电极(曲线d)的EIS半径远大于非印迹电极(曲线c)的EIS半径,这是由于草甘膦是非电活性物质,被掺杂于聚吡咯骨架中阻碍了电子的传递,其阻抗增加。而洗脱模板分子后电极留下的“孔穴”使部分电子探针到达电极表面,所以洗脱后印迹电极(曲线b)的EIS半径小于非印迹电极的半径,表明其电子转移阻抗比非印迹电极阻抗小。
2.2 实验条件的优化
2.2.1 检测体系及pH值的影响 分别配制含1 μg/mL草甘膦的各种溶液(BR缓冲液、PBS缓冲液和甲酸水溶液),各溶液pH值为2.0,3.0,4.0,5.0,6.0,7.0,将印迹电极在相同条件下进行吸附检测,结果表明印迹电极在含草甘膦的甲酸水溶液中检测信号最高,在其他溶液中对草甘膦基本无响应。因此,选择甲酸水溶液作为检测草甘膦的溶液体系。由于不同的pH值条件下,溶液中草甘膦所带的电荷不同,电极对草甘膦的响应也不同。因此本实验研究了印迹电极在不同pH值(3.0,4.0,5.0,6.0,7.0)甲酸水溶液中对草甘膦的响应。结果表明,当溶液的pH值为4.0时,印迹电极对草甘膦的吸附最大,其探针分子的电流值最小;随着体系pH值增大,印迹电极对草甘膦的吸附减小,其探针分子的响应电流值增大,说明pH值对印迹电极检测草甘膦具有重要的影响,推测可能是随着pH值增加,溶液中的草甘膦分子带负电荷增多,与过氧化聚吡咯膜带负电的羰基和羧基产生静电排斥,从而导致吸附减少,探针分子电流增大[22]。因此,实验选择pH 4.0作为检测草甘膦的最佳酸碱度。
2.2.2 吸附时间的影响 采用差分脉冲伏安法(DPV)考察了50 ng/mL草甘膦在洗脱模板后的分子印迹电极上峰电流随时间的变化情况。结果显示,随着吸附时间的延长,印迹电极对草甘膦的吸附量增加,其探针分子的峰电流逐渐降低,当18 min之后峰电流几乎不变。因此,选择18 min作为检测体系的最佳吸附时间。

图6 印迹电极的吸附和洗脱效果Fig.6 Absorption and elution of imprinted electrode towards glyphosatea:NIP-modified gold electrode after elution;b:MIP-modified gold electrode after elution;c:NIP-modified gold electrode after absorption;d:MIP-modified gold electrode after absorption
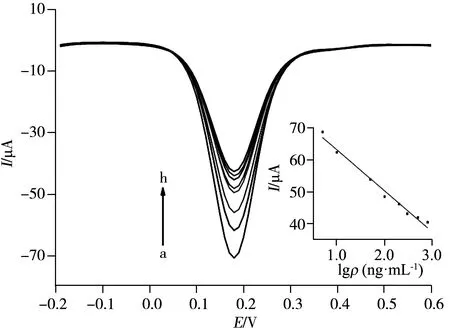
图7 吸附不同浓度草甘膦的分子印迹传感器在铁氰化钾溶液中的DPV图Fig.7 DPV curves of MIP-modified gold electrode in different concentration of Gly concentration of Gly(a-h):5,10,50,100,200,300,500,800 ng/mL;5.0 mmol/L K3[Fe(CN)6],0.1 mol/L KCl;insert:calibration curve of glyphosate
2.3 印迹电极与非印迹电极吸附的对比
将印迹电极与非印迹电极在相同条件下吸附50 ng/mL草甘膦18 min后,观察吸附和洗脱模板分子前后DPV的变化(图6)。结果显示,非印迹电极对草甘膦的响应微小,而印迹电极由于吸附了草甘膦其探针分子的电流发生了很大变化,当模板分子洗脱后,印迹电极的探针响应电流又恢复,说明印迹电极对草甘膦具有特异性吸附性能,也表明了洗脱条件的效果。
2.4 方法的线性范围与检出限
采用差分脉冲伏安法(DPV)考察了分子印迹电极吸附不同浓度的草甘膦18 min后的峰电流变化趋势(图7),草甘膦浓度(ρ,ng/mL)在5~800 ng/mL范围内与峰电流(I)呈良好的线性关系,线性方程为I=-12.86lgρ+75.92,r2=0.981 7,检出限(S/N=3)为0.27 ng/mL。
2.5 电极的选择性
为研究修饰电极对草甘膦的选择性,采用差分脉冲伏安法测定印迹传感器和非印迹传感器对1 000 ng/mL草甘膦、氨甲基膦酸(AMPA)、毒死蜱(Chlorpyrifos)、涕灭威(Aldicarb)的吸附能力。实验结果表明,印迹电极对草甘膦的吸附最大,对毒死蜱和氨甲基膦酸有少量吸附,这可能是印迹识别位点与磷酸根有关,而对于结构差异较大的涕灭威几乎不吸附;非印迹电极对以上农药几乎无吸附,这说明印迹传感器对于草甘膦有很好的特异性识别性能,尽管对以上结构类似物有少量吸附,但不影响传感器对草甘膦的检测。
2.6 电极的重现性与稳定性
在相同条件下制备5支印迹电极,对50 ng/mL 草甘膦溶液平行测定5次,计算电流响应值的相对标准偏差为3.1%,表明该印迹传感器具有良好的重现性,分子印迹电极经洗脱可重复利用,表明模板分子与印迹膜上“孔穴”的结合为可逆过程,印迹电极可重复使用。电极经多次使用,洗脱后置于超纯水中常温保存,3周后其响应值降至初始响应值的80.9%,表明印迹电极的稳定性良好。
2.7 实际样品的加标检测
为了评价本方法的实用性,随机选择9份自来水进行加标回收率实验。分别取100,200,500 μL的1 μg/mL草甘膦标准溶液用自来水定容至10 mL,加标浓度分别为10,20,50 ng/mL。不做任何前处理,直接用分子印迹电极检测18 min,每个样品平行测定3次。结果测得草甘膦在自来水样品中的回收率为78.6%~99.0%,相对标准偏差为2.8%~4.5%,表明草甘膦分子印迹电化学传感器可以实现自来水中草甘膦农药残留的快速检测。
3 结 论
本文以吡咯为功能单体,草甘膦为模板分子,通过电化学聚合和聚吡咯过氧化法,在电极表面合成了聚吡咯多孔印迹膜,制备了草甘膦分子印迹电化学传感器。该传感器制备方便,模板分子掺杂和洗脱简单,对目标物的特异性强、稳定性好、传质速度快、响应时间短(仅为18 min)。在5~800 ng/mL草甘膦浓度范围内,传感器响应值与草甘膦浓度的对数值呈良好的线性关系,其检出限为0.27 ng/mL,对结构类似物等干扰物质具有很强的抗干扰能力,可满足饮水中痕量草甘膦的检测要求。
[1] Duke S O,Powles S B.PestManag.Sci.,2008,64(4):319-325.
[2] Guyton K Z,Loomis D,Grosse Y,El Ghissassi F,Benbrahim-Tallaa L,Guha N,Scoccianti C,Mattock H,Straif K.LancetOncol.,2015,16(5):490-491.
[3] Horciciak M,Masar M,Bodor R,Danc L,Bel P.J.Sep.Sci.,2012,35(5/6):674-680.
[4] Coutinho C F B,Coutinho L F M,Mazo L H,Nixdorf S L,Camara C A P.J.Chromatogr.A,2008,1208(1/2):246-249.[5] Popp M,Hann S,Mentler A,Fuerhacker M,Stingeder G,Koellensperger G.Anal.Bioanal.Chem.,2008,391(2):695-699.
[6] GB 5749-2006.Standards for Drinking Water Quality.National Standards of the People's Republic of China(生活饮用水卫生标准.中华人民共和国国家标准).
[7] Fang F,Xu H,Wei R Q,Liu X N,Xu R,Li S C,Liu B K.J.Instrum.Anal.(方芳,徐会,魏荣卿,刘晓宁,徐蓉,李寿椿,刘宝菎.分析测试学报),2011,30(6):683-686.
[8] Zhou S,Xu D M,Lin L Y,Chen L P,Zhou Y,Yang L Z.J.Instrum.Anal.(周爽,徐敦明,林立毅,陈鹭平,周昱,杨黎忠.分析测试学报),2013,32(2):199-204.
[9] Steinborn A,Alder L,Michalski B,Zomer P,Bendig P,Martinez S A,Mol H G J,Class T J,Costa P N.J.Agric.FoodChem.,2016,64(6):1414-1421.
[10] Lanaro R,Costa J L,Cazenave S O S,Zanolli-Filho L A,Tavares M F M,Chasin A A M.J.ForensicSci.,2015,60(S1):241-247.
[11] Zhang P Z,Wu J,Zhang P M,Xu Y.J.Instrum.Anal.(张培志,吴军,张培敏,徐育.分析测试学报),2003,22(4):89-90.
[12] Selvi A A,Sreenivasa M A,Manonmani H K.FoodAgric.Immunol.,2011,22(3):217-228.
[13] Cai Q,Chen X,Qiu B,Lin Z.Chin.J.Chem.,2011,29(3):581-586.
[14] Yao T,Gu X,Li T F,L J G,L J,Zhao Z,Wang J,Qin Y C,She Y X.Biosens.Bioelectron.,2016,75:96-100.[15] She Y X,Cao W Q,Shi X M,Lv X L,Liu J J,Wang R Y,Jin F,Wang J,Xiao H.J.Chromatogr.B,2010,878(23):2047-2053.
[16] Wong A,Sotomayor M D P T.J.Electroanal.Chem.,2014,731:163-171.
[17] Toro M J U,Marestoni L D,Sotomayor M D P T.Sens.ActuatorB,2015,(208):299-306.
[18] Li X,Zhang L M,Wu C R,Wei X P,Li J P.J.Instrum.Anal.(李雪,张连明,吴昌儒,魏小平,李建平.分析测试学报),2013,32(11):1344-1348.
[19] Pan M F,Fang G Z,Liu B,Qian K,Wang S.Anal.Chim.Acta,2011,690(2):175-181.
[20] Sahin M,Sahin Y,Ozcan A.Sens.ActuatorB,2008,133:5-14.
[21] Susana M,Otto S W.Anal.Chim.Acta,1996,334:149-153.
[22] McConnell J S,Hossner L R.J.Agric.FoodChem.,1985,6(33):1075-1078.
Preparation of an Electrochemical Sensor Based on Molecularly Imprinted Polymer and Its Application in Determination of Glyphosate Residues in Water Samples
ZHANG Chao1,LI Teng-fei1,ZHAO Feng-nian1,WANG Shan-shan1,SHE Yong-xin1*,JIN Mao-jun1,LIU Hai-jin2,JIN Fen1,SHAO Hua1,ZHENG Lu-fei1,XU Ping2,WANG Jing1*
(1.Institute of Quality Standards&Testing Technology for Agri-Products,Chinese Academy of Agricultural Sciences,Beijing 100081,China;2.Tibet Testing Center of Quality and Safety for Agricultural and Animal Husbandry Products,Lhasa 850000,China)
A novel ultra-sensitive molecularly imprinted electrochemical senor with good imprinted capacity to glyphosate was prepared by electropolymerisation on the gold electrode surface,with pyrrole(Py) as functional monomer,glyphosate(Gly) as template.The prepared electrode was characterized by cyclic voltammetry(CV),differential pulse voltammetry(DPV) and electrochemical impedance spectroscopy(EIS).The condition of polymerization,the method of eluting template,the pH value of detection andincubation time were optimized.The optimal conditions were as follows:supporting electrolyte:Britton Robison buffer solution(BR,pH 5.0),ratio of template to functional monomers:1∶5,incubation system:formic acid aqueous solution(pH 4.0),incubation time:18 min.Under the optimum experimental conditions,the prepared electrode showed rapid response,high sensitivity and good selectivity for the template molecule glyphosate.A good linear relationship between oxidation peak current and Gly concentration was obtained over the range of 5-800 ng/mL with a correlation coefficient of 0.981 7 and a detection limit(S/N=3) of 0.27 ng/mL.The prepared sensor also showed good reproducibility and stability,the sensor was successfully applied in the determination of glyphosate in the tap water with recoveries of 78.6%-99.0%.
pyrrole;glyphosate;molecularly imprinted electrochemical sensor;tap water
2016-05-19;
2016-06-30
国家自然科学基金项目(31471654);“十二五”国家科技支撑计划项目( 2014BAD13B05-05)
10.3969/j.issn.1004-4957.2016.12.005
O657.1;S482.4
A
1004-4957(2016)12-1542-06
*通讯作者:佘永新,研究员,研究方向:仿生识别材料与检测技术,Tel:010-82106513,E-mail:0891syx@163.com 王 静,教授,研究方向:仿生识别材料与检测技术,Tel:010-82106568,E-mail:wjing_2001@163.com
猜你喜欢
中学生数理化·中考版(2022年12期)2022-02-16
四川轻化工大学学报(自然科学版)(2021年3期)2021-08-30
农药科学与管理(2019年6期)2019-11-23
世界农药(2019年2期)2019-01-06
电子制作(2016年23期)2016-05-17
肇庆学院学报(2016年5期)2016-03-11
合成化学(2015年10期)2016-01-17
环境科技(2015年4期)2015-11-08
营销界(2015年23期)2015-02-28
华东师范大学学报(自然科学版)(2014年4期)2014-03-11
