
重庆两株H5N1流感病毒的HA、NA基因克隆及序列分析
2017-02-09 02:06胡仁建蔡家利刘海军李重阳王冬梅
重庆理工大学学报(自然科学) 2017年1期
胡仁建, 顾 盼, 蔡家利, 刘海军,丁 森,张 奎, 李重阳, 陈 华,王冬梅
(1.重庆理工大学 药学与生物工程学院, 重庆 400074;2.西南大学 家蚕基因组生物学国家重点实验室, 重庆 400074;3.重庆市公安局, 重庆 400074; 4.南京大学, 南京 210023;5.珠海联邦制药有限公司, 广州 528400; 6.中国农业大学, 北京 100193)
重庆两株H5N1流感病毒的HA、NA基因克隆及序列分析
胡仁建1,2, 顾 盼1, 蔡家利1, 刘海军3,丁 森4,张 奎2, 李重阳2, 陈 华5,王冬梅6
(1.重庆理工大学 药学与生物工程学院, 重庆 400074;2.西南大学 家蚕基因组生物学国家重点实验室, 重庆 400074;3.重庆市公安局, 重庆 400074; 4.南京大学, 南京 210023;5.珠海联邦制药有限公司, 广州 528400; 6.中国农业大学, 北京 100193)
2012年,从重庆北碚区和巴南区的家禽养殖场死鸭和病鸡体内分离出两株H5N1禽流感病毒,分别命名为A/duck/Chongqing/BB/H5N1和A/chicken/Chongqing/BN/H5N1,并对这两株H5N1禽流感病毒的HA、NA和M蛋白的基因进行了克隆及序列分析,在基因和蛋白质水平上分析其同源性。遗传进化树分析结果表明:这两毒株的HA、NA和M基因不处于同一支上,亲缘关系较远;这两毒株的HA、NA、M蛋白的基因同源性分别达95%,95%,97% ;两毒株的HA蛋白裂解位点氨基酸序列均为RERRR-KR,符合高致病性禽流感的特征。本次分离的流感病毒的基因克隆鉴定和序列分析为禽流感的流行病学研究、疫苗研制以及防控提供重要理论依据。
禽流感病毒;H5N1;基因克隆;序列分析;进化分析
高致病性禽流感(high pathogenic avian influenza,HPAI)是由A型流感病毒引起家禽高致病性高死亡率的疾病。该病毒分布广泛,国际兽疫局将其划定为A类传染病,是世界各国检疫和防控的重点对象[1-2]。HPAI病毒可在家禽中传播流行,引起大规模爆发,导致世界范围内禽类副产品的严重下降以及禽类的大量死亡。1996年,在中国广东省的家鹅中分离鉴定出第1株高致病性H5N1病毒(A/goose/Guangdong/1/96)[3]。该毒株被认为是H5N1病毒的祖先毒株,现流行的H5N1病毒多是由该原始毒株演化而来的[3-5]。自1996年以来,祖先毒株以不可预测的方式快速蔓延至亚洲、欧洲及非洲的许多国家[6]。在物种间持续的传播流行使得H5N1病毒在不同的地理区域基因分化形成进化亲缘关系较远的各个分支。
HA和NA是流感病毒粒子表达最丰富的两种表面抗原,它们的基因变异也是最为频繁的[7-8]。HA 是决定病毒致病性、毒力强弱和宿主特异性等方面的关键因素之一,NA在变异中起次要作用[9-10]。此外,高致病力毒株的 HA 在其裂解位点附近均有多个碱性氨基酸,而低致病力毒株的HA 在其裂解位点只有一个精氨酸(R)[5]。本文对2012年从重庆发病地区病死鸭和病鸡体内中分离鉴定出2株H5N1亚型AIV进行HA和NA基因克隆和序列分析,探讨2株H5N1亚型AIV的变异、进化特点、HA蛋白的裂解位点的差异,为禽流感的有效防控提供参考依据。
1 材料与方法
1.1 主要试剂与仪器
病死鸭和病鸡分别来自重庆市北碚区一鸭场和重庆市巴南区一鸡场;SPF级鸡胚购自重庆长寿区特驱有限公司;TIANamp Virus DNA/RNA Kit购于天根生化科技(北京)有限公司;质粒抽提试剂盒购自Omega公司;PrimeScript®RT reagent Kit、T19载体和JM109感受态细胞购自宝生物(大连)公司;引物由宝生物(大连)公司合成;Thermo ScientificTMPCR 仪购自Thermo Scientific公司;禽流感病毒H5N1、H7N1和H9N2标准血清以及鸡新城疫标准血清由重庆理工大学药学与生物工程学院生物制药实验室保存。
无菌采集死鸭、病鸡的肺、脾、肾、肝、脑等病料,用液氮冷冻磨碎病料后加入无菌0.9%生理盐水,10 000 r/min离心30 min,用0.45 μm和0.22 μm过滤取上清液。按尿囊腔法接种9日龄的SPF级鸡胚,死鸭和病鸡的处理病料液分别以200 μL/只接种鸡胚,注射无菌生理盐水作为空白对照。37℃孵育,每天检查并收集24 h后死亡鸡胚的尿囊液。
1.3 尿囊液的红细胞凝集试验和红细胞凝集抑制试验
按照参考文献[11]做红细胞凝集试验,测定用0.5%甲醛灭活后的鸡胚尿囊液的病毒血凝效价,用标准的禽流感病毒H5N1、H7N1、H9N1和新城疫病毒血清做红细胞血凝抑制试验。
1.4 阳性尿囊液禽流感病毒HA、NA和M基因的克隆和测序
为进一步鉴定两分离毒株亚型,引用中华人民共和国农业行业标准(NY/T772—2004)中禽流感病毒RT-PCR实验方法中的引物6对:引物M-229U/M-229L是特异性扩增M基因的,以此来确定其属于A类禽流感[12-13];引物H5-380U/H5-380L、 H7-501U/H7-501L 和H9-732U/H9-732L可分别扩增H5、H7和H9基因;N1和N2基因的特异性引物则分别为N1-358U/N1-358L 和 N2-377U/N2-377L。根据RNA提取试剂盒说明书,提取血清学鉴定为阳性的尿囊液的RNA。将RNA反转录为cDNA,以cDNA为模板PCR扩增H5、H7、H9、N1、N2和M基因产物。PCR的反应体系是50 μL,包含双蒸灭菌水37.5 μL、反转录产物4 μL、上下游引物各0.5 μL、PCR Buffer 5.0 μL、2.5 mmol/L的dNTPs 2.0 μL、Taq聚合酶 0.5 μL。PCR反应条件:95 ℃预变性5 min;94 ℃变性45 s,52 ℃退火45 s,72 ℃延伸45 s,循环30次;72 ℃再延伸6 min。同时设置阳性和阴性对照。DNA电泳鉴定PCR产物。将PCR产物H5、N1、M基因分别与19-T载体连接并转化入感受态细胞JM109中,将经 PCR鉴定为阳性克隆质粒的送宝生物(大连)公司测序。
1.5 基因比对、进化树的构建和蛋白质序列分析
将从禽类中分离两个毒株的H5、N1、M基因的核酸序列提交至Clustal W进行多序列比对,氨基酸的残基分析利用BioEdit 7.1.5软件。利用NCBI对相应序列进行BLAST,分析这两毒株之间的同源性等,并与 GenBank中其他HA基因的核酸序列进行比对,分析两毒株与其他参考毒株的同源性。病毒的分支鉴定参考WHO/FAO H5N1 Evolution Working Group[14-15]。利用MEGA5.05软件采用邻接法(neighbor-joining method,NJ)构建遗传进化树,以序列的同源性、地域来源、分离时间以及WHO和FAO推荐的的代表性H5N1毒株为基准。
消除方法的基本思路是将定向耦合器3端口和4端口的测量数据均等效为被测发射机端口的发射数据,然后进行对消.
2 结果
2.1 病毒的分离与鉴定
接种北碚死鸭病料处理液的鸡胚在36~64 h死亡,接种巴南病鸡处理液的鸡胚在48~ 56 h死亡,接种生理盐水的对照组未死亡。所有死亡鸡胚全身出血,接种北碚死鸭病料处理液的鸡胚比接种巴南病鸡处理液的鸡胚出血更严重,所有死亡鸡胚的体型小于空白对照鸡胚,死亡鸡胚尿囊液都清晰红色透明,空白鸡胚尿囊液无色透明。
红细胞凝集试验显示:北碚死鸭病料处理液的鸡胚和接种巴南病鸡处理液的鸡胚的尿囊液的血凝效价分别为1∶128和1∶32。红细胞凝集抑制试验显示:H5N1和H7N1标准血清均可抑制两毒株,其血凝抑制效价均为1∶32和1∶8。标准的禽流感病毒H9N1病毒血清和新城疫病毒血清均不能抑制两株病毒。
如图1(a)所示: RT-PCR扩增出与禽流感病毒H5(380bp)、N1(358bp)基因大小一致的PCR产物片段,阴性对照无对应的产物,说明两株禽流感病毒的亚型为H5N1;如图1(b)所示: RT-PCR扩增出与禽流感病毒M(229bp)基因大小一致的PCR产物片段,阴性对照无对应的产物,提示两毒株均为A类禽流感病毒。分别将两毒株命名为A/duck/Chongqing/BB/2012(简称北碚株) 和A/chicken/Chongqing/ BN/2012(简称巴南株)。
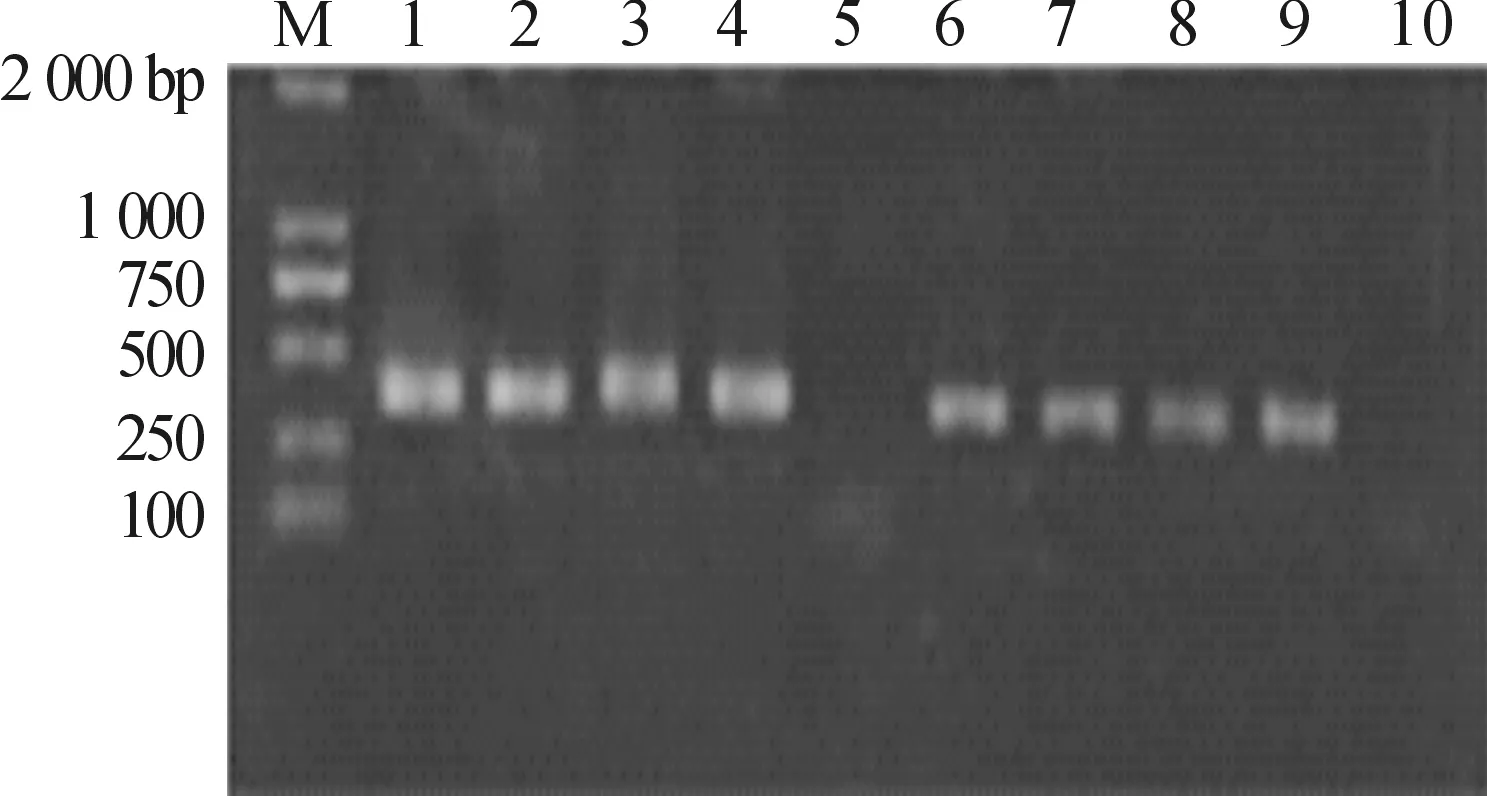
M,DL2000Marker;1,2.北碚株 H5 PCR产物; 3,4.巴南株 H5 PCR产物; 5.H5 PCR阴性对照; 6,-7.北碚株 N1 PCR产物; 8,9.巴南株 N1 PCR产物;10.N1 PCR阴性对照。
(a)H5和N1基因 PCR产物鉴定
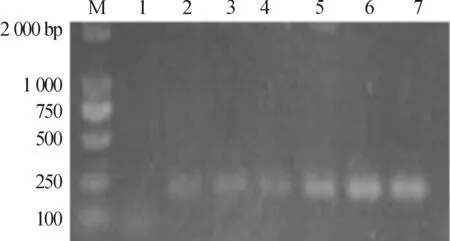
M, DL2000Marker;1,M基因阴性对照;2,3,4.北碚株 M PCR产物;5,6,7.巴南株 M PCR产物。
(b)M基因 PCR产物鉴定
图1 PCR产物鉴定
Fig.1 PCR product identification
2.2 北碚株和巴南株的HA及NA基因的核酸序列比对
将北碚株和巴南株的HA、NA和M基因的核酸序列共6个上传至NCBI,获得序列号为:BankIt1936040 Seq1 KX544798,BankIt1936175 Seq2 KX544799,BankIt1936181 Seq3 KX544800,BankIt1936183 Seq4 KX544801,BankIt1936185 Seq5 KX544802,BankIt1936188 Seq6 KX544803。两毒株核酸序列比对显示:北碚株与巴南株之间HA和NA基因均存在17个碱基的差异,且它们的HA和NA基因的核酸同源性均为95%;此外,它们的M基因有7个碱基不同,同源性达97%。
2.3 遗传进化树分析
为了更好地理解两分离毒株的进化历史,构建了HA、NA和M基因的遗传进化树。如图2(a)所示,可见北碚株和巴南株的HA基因分别属于clade2.3.2.1及clade2.3.4.2。从HA基因进化树上可以看出,北碚株的HA核酸序列与A/duck/Hunan/8/2008的同源性最高,达98%,而巴南株的HA核酸序列与A/environment/Guizhou/4/2009有99%的同源性。发现巴南株和北碚株与A/bar-headed goose/Qinghai/3/2005(H5N1)和A/bar-headed goose/Tibet/8/2006(H5N1)在同一大支,推测分离的两株H5N1病毒株可能来源于后两株。这可能是H5N1禽流感病毒进出南方的佐证,病毒很有可能是通过野鸟的迁徙传播的。
如图2(b)所示:北碚株和巴南株的NA基因不在同一个分支上,亲缘关系较远。北碚株与代表性的参考毒株A/duck/Hunan/8/2008和A/Hubei/1/2010处于同一支上。北碚株与巴南株的NA核酸序列分别与A/wild duck/Fujian/1/2011 和A/environment/Guizhou/4/2009均有99%的同源性。
如图2(c)所示:北碚株与巴南株的M核酸序列分别与A/duck/Wenzhou/YHQL22 /2014(H5N6)和A/environment/Guizhou /4/2009毒株的同源性最高,均达99%。
2.4 蛋白变异与裂解位点分析
如表1所示:在HA蛋白比对中,两株毒株之间存在两个氨基酸的替换,它们HA蛋白的同源性达98%。两毒株的H5蛋白裂解位点一致,均为RRRKR,为5个连续的碱性氨基酸序列,符合高致病性禽流感的特征,但与祖先毒株A/goose/Guangdong/1/96 相比,均缺失一个碱性氨基酸残基赖氨酸(K)。与祖先毒株A/goose/Guangdong/1/96相比,两毒株均存在抗原漂移现象:HA蛋白中,巴南株为298M-I、326R-K、336T-S和338Q-L,北碚株为293K-R、326R-K、336T-S和338Q-L;NA蛋白中,巴南株有259E-K和289T-I两个氨基酸突变,北碚株有232A-T、258M-I和289T-I三个氨基酸突变;M蛋白中,两毒株均有15V-I和27R-K两个氨基酸突变(见表2)。两毒株的M蛋白氨基酸序列一致,均与A/chicken/Hubei/wn/2003毒株的M蛋白有最高的同源性(见表2)。
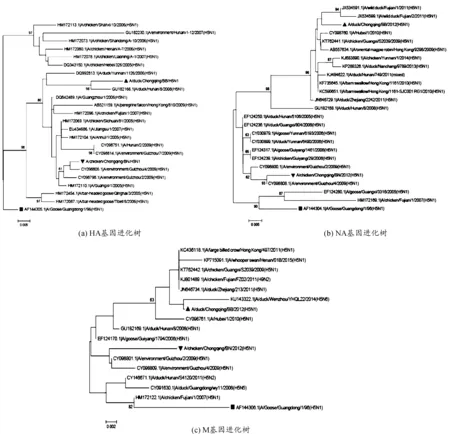
图2 基因进化树 Fig.2 Phylogenetic trees from neighbor-joining analyses表1 分离株与中国其他毒株HA蛋白氨基酸比对及裂解位点分析Table 1 HA amino acid subsititution comparison between studied strains and other Chinese isolates.
H5蛋白氨基酸序号是根据A/goose/Guangdong/1/1996。
表2 分离株与中国其他毒株NA、M蛋白氨基酸比对Table 2 Deduced amino acid sequences of NA and M1 in the BB and BN strains relative to some genetically related strains
NA、M蛋白氨基酸序号是根据A/goose/Guangdong/1/1996。
3 讨论
1996年H5N1亚型禽流感病毒首次在中国广东省病鹅中检测到,分为无致病性和高致病性两种,自H5N1首次被鉴定后,在中国20多个省的多个农场又出现H5N1大流行[1-2,16]。高致病性H5N1病毒clade2.3.2首次在2005年从中国以及越南的鸭、鹅中鉴定得到[17- 18]。从2006年以来,在香港从野鸟中分离的H5N1病毒中,clade2.3.2和clade2.3.4占据主导地位[19-0]。FAO-OIE-WHO表示clade2.3.2.1在东南亚的流行是目前H5N1频繁爆发的主要原因,目前在印度、老挝、越南、尼泊尔、中国、俄罗斯、日本和孟加拉均有H5N1clade2.3.2.1病毒株的相关报道[13,21-24]。此外,从2005年以来,由H5N1clade2.3.4病毒株的H5基因变异的新型毒株在中国以及其他地区传播流行[25]。
在本试验中,从重庆市北碚区某鸭场和巴南区某鸡场分离出两种病毒株。血清学研究结果表明:H5及H7标准血清可抑制两种病毒。通过RT-PCR进行进一步鉴定,M基因RT-PCR结果表明两种病毒均属于禽流感A类病毒,并且均可扩增出H5和N1的特异性条带,将其命名为A/duck/Chongqing/BB/2012(北碚株) 和A/chicken/Chongqing/BN/2012(巴南株)。进化分析结果表明:北碚株和巴南株的HA基因分别属于clade2.3.2.1及clade2.3.4.2。通过进化树分析发现:巴南株的HA,NA,M基因均与A/environment/Guizhou /4/2009有较高的同源性,而北碚株则与A/duck/Hunan/8/2008 和A/Hubei/1/2010接近。此外,两毒株的H5蛋白氨基酸裂解位点序列均为RERRR-KR,符合高致病禽流感的特征。裂解位点缺失一个K,可能对病毒毒力有一定影响。M.S.Lee等[6]发现:含RERRR-KR位点的Dk/CHN/E319-2/03病毒株对小鼠进行攻毒试验时,其致病性实质上是弱于从1997年到2006年对人致死的案例中分离得到的H5N1病毒(RERRRKKR)。从这一点看,这可能也是这两株病毒株仅引起小范围流行而非大规模爆发的原因。北碚株在发病时流感症状典型严重,在本鸭场传播速度很快,抗病毒、抗菌治疗和再次注射禽流感疫苗和新城疫疫苗无效,在7~14天的短时间内所有鸭子全部死亡,造成了巨大的经济损失;而巴南株在鸡场发病时流感症状不典型,抗病毒、抗菌治疗和再次注射禽流感疫苗和新城疫疫苗有效控制了病情,大部分鸡恢复健康,死亡的鸡数目较少。从以上临床症状反映出北碚株的毒力大于巴南株,从进化树结果可以发现,北碚株的H5、N1和M基因均比巴南株进化的快,提示抗原漂移在其中起到了重要作用。但由于未对H5、N1和M基因全长进行测序,北碚株及巴南株毒力差异的原因仍不是完全清楚。
本研究全面分析了中国西南的中心——重庆爆发的两株H5N1禽流感病毒株——A/duck/Chongqing/BB/2012和A/chicken/Chongqing/BN/2012,探讨了这两株毒株的变异及进化特征,在一定程度上为H5N1禽流感在中国南方的进化提供参考信息。笔者推测:H5N1的clade2.3.2和clade2.3.4这两支可能仍在中国南方流行并占据主导地位,这也是人类健康的一大隐患。另一方面,我国从2004年开始通过疫苗接种的手段预防H5N1禽流感病毒[26-,27],尽管这一手段有效降低了H5N1在禽类中的发病率并显著减少人感染的案例数,但是,由于禽类基数的庞大,难以做到对每只野鸟都进行免疫,H5N1型禽流感仍以减缓的频率在中国南方持续爆发[26]。因此,应该清楚地认识到对H5N1在禽类中的持续监测以及有效的控制手段的不断更新是迫切需要的。此外,由于H5N1变异株的不断出现及跨区域的持续传播,区域性的有效防控措施必须摆在重要、突出的位置上[10]。而只有整合实时的病毒学信息、通过快速诊断方法获取的基因信息以及配套的严格控制质量的疫苗产品,才能真正做到系统地有效防控H5N1的发生及流行。本实验中A/duck/Chongqing/BB/H5N1(2012)和 A/chicken/Chongqing/BN/H5N1(2012)两毒株的分离与鉴定,HA、NA和M基因的克隆与序列分析,为禽流感病毒H5N1的流行病学研究(包括变异分析、进化分析)和疫苗的研制提供更多的信息,对流感的防控具有重要意义。
[1] LI Y,SHI J,ZHONG G,et al.Continued evolution of H5N1 influenza viruses in wild birds,domestic poultry,and humans in China from 2004 to 2009[J].J Virol,2010,84(17):8389-97.
[2] REJMANEK D,HOSSEINI P R,MAZET J A,et al.Evolutionary Dynamics and Global Diversity of Influenza A Virus[J].J Virol,2015,89(21):10993-1001.
[3] XU Xiyan,NANCY J C,GUO Yuanji.Genetic Characterization of the Pathogenic Influenza A/Goose/Guangdong/1/96 (H5N1) Virus:Similarity of Its Hemagglutinin Gene to Those of H5N1 Viruses from the 1997 Outbreaks in Hong Kong[J].Virology,1999,261:15-19.
[4] ALKHAMIS M A,MOORE B R,PEREZ A M.Phylodynamics of H5N1 Highly Pathogenic Avian Influenza in Europe,2005—2010:Potential for Molecular Surveillance of New Outbreaks[J].Viruses,2015,7(6):3310-3328.
[5] WAN X F,REN T,LUO K J,et al.Genetic characterization of H5N1 avian influenza viruses isolated in southern China during the 2003-04 avian influenza outbreaks[J].Arch Virol,2005,150(6):1257-1266.
[6] LEE M S,DENG M C,LIN Y J,et al.Characterization of an H5N1 avian influenza virus from Taiwan[J].Vet Microbiol,2007,124(3/4):193-201.
[7] EICHELBERGER M C,WAN H.Influenza Neuraminidase as a Vaccine Antigen[J].Springer International Publishing,2014,386:275-299.
[8] BOTTCHER-FRIEBERTSHAUSER E,GARTEN W,MATROSOVICH M,et al.The hemagglutinin:a determinant of pathogenicity[J].Curr Top Microbiol Immunol,2014,385:30-34.
[9] GARCIA J M,LAI J C C,HASELHORST T,et al.Investigation of the binding and cleavage characteristics of N1 neuraminidases from avian,seasonal,and pandemic influenza viruses using saturation transfer difference nuclear magnetic resonance[J].Influenza and Other Respiratory Viruses,2014,8(2):235-242.
[10]SMITH G J,FAN X H,WANG J,et al.Emergence and predominance of an H5N1 influenza variant in China[J].Proc Natl Acad Sci U S A,2006,103(45):16936-16941.
[11]中华人民共和国农业部.中华人民共和国兽用生物制品质量标准二00一年版[S].
Ministry of Agriculture of the People’s Republic of China.People’s Republic of China veterinary biological products quality standards two edition of 2001[S].
[12]RON A M, FOUCHIE T M B,SANDER H.Detection of Influenza A Viruses from Different Species by PCR Amplification of Conserved Sequences in the Matrix Gene[J].journal of clinical microbiology,2000,38(11):4096-4101.
[13]PARVIN R,KAMAL A H,HAQUE M E,et al.Genetic characterization of highly pathogenic H5N1 avian influenza virus from live migratory birds in Bangladesh[J].Virus Genes,2014,49(3):438-48.
[14]GROUP W O.Continued evolution of highly pathogenic avian influenza A (H5N1):updated nomenclature[J].Influenza Other Respir Viruses,2012,6(1):1-5.
[15]World Health Organization/World Organisation for Animal H F,Agriculture Organization H N E W G.Revised and updated nomenclature for highly pathogenic avian influenza A (H5N1) viruses[J].Influenza Other Respir Viruses,2014,8(3):384-8.
[16]SU S,BI Y,WONG G,et al.Epidemiology,Evolution,and Recent Outbreaks of Avian Influenza Virus in China.[J].Journal of Virology,2015,89(17):8671-8676.
[17]ROBERTON S I,BELL D J,SMITH G J,et al.Avian influenza H5N1 in viverrids:implications for wildlife health and conservation.[J].Proceedings Biological Sciences,2006,273(1595):1729-1732.
[18] CHEN H,SMITH G J,LI K S,et al.Establishment of multiple sublineages of H5N1 influenza virus in Asia:implications for pandemic control.[J].Proceedings of the National Academy of Sciences,2006,103(103):2845-2850.
[19]SMITH G J D,VIJAYKRISHNA D,ELLIS T M,et al.Characterization of avian influenza viruses A (H5N1) from wild birds,Hong Kong,2004—2008.[J].Emerging Infectious Diseases,2009,15(3):402-407.
[20]ELLIS T M,DYRTING K C,WONG C W,et al.Analysis of H5N1 avian influenza infections from wild bird surveillance in Hong Kong from January 2006 to October 2007[J].2009,38(2):107-119.
[21]NAGARAJAN S,TOSH C,SMITH D K,et al.Avian influenza (H5N1) virus of clade 2.3.2 in domestic poultry in India[J].Plos One,2012,7(2):e31844.
[22]KIM B S,KANG H M,CHOI J G,et al.Characterization of the low-pathogenic H5N1 avian influenza virus in South Korea[J].Poult Sci,2011,90(7):1449-1461.
[23]HARTANINGSIH N,WIBAWA H,PUDJIATMOKO,et al.Surveillance at the molecular level:Developing an integrated network for detecting variation in avian influenza viruses in Indonesia[J].Prev Vet Med,2015,120(1):96-105.
[24]NGUYEN D T,BRYANT J E,DAVIS C T,et al.Prevalence and distribution of avian influenza a(H5N1) virus clade variants in live bird markets of Vietnam,2011—2013[J].Avian Dis,2014,58(4):599-608.
[25]SMITH G J,DONIS R O.Nomenclature updates resulting from the evolution of avian influenza A(H5) virus clades 2.1.3.2a,2.2.1,and 2.3.4 during 2013—2014[J].Influenza Other Respir Viruses,2015,1750-2659.[26]SMITH G J D,FAN X H,WANG J,et al.Emergence and predominance of an H5N1 influenza variant in China.[J].Proceedings of the National Academy of Sciences of the United States of America,2006,103(45):16936-16941.[27]LI C,BU Z,CHEN H.Avian influenza vaccines against H5N1 “bird flu”.[J].Trends in Biotechnology,2014,32(3):147-156.
(责任编辑 刘 舸)
HA and NA Gene Clone and Sequence Analysis of Two H5N1 Virus Isolates from Chongqing
HU Ren-jian1,2, GU Pan1, CAI Jia-li1, LIU Hai-jun3, DING Sen4,ZHANG Kui2, LI Chong-yang2, CHEN Hua5, WANG Dong-mei6
(1.College of Pharmacy and Biological Engineering, Chongqing University of Technology,Chongqing 400074, China; 2.State Key Laboratory of Silkworm Genome Biology,Southwestern University, Chongqing 400074, China; 3.Chongqing City Public Security Bureau, Chongqing 400082, China; 4.Nanjing University, Nanjing 210023, China;5. Zhuhai United Laboratories Co. Ltd.,Guangzhou 528400, China;6.China Agricultural University, Beijing 100193, China)
Avian influenza (AI), a virus subtype H5N1 virus is a highly pathogenic avian influenza (HPAI), which is panzootic in poultry, then continues to spread and pose a major challenge to animal and human health. In 2012, two outbreaks of HPAI virus occurred and we isolated two H5N1 virus strains from two poultry farms in the Chongqing Beibei District and Banan District, named A/duck/Chongqing/BB/2012 and A/chicken/Chongqing/BN/2012, respectively. In this study, the hemagglutinin (HA), neuraminidase (NA) and matrix protein (M) genes of these two strains were cloned and sequenced, and then the homogeneity and the heterogeneity were analyzed at the genetic and proteinic levels. The results shows that the homogeneity of the HA, NA and M genes was 95%, 95% and 97%, respectively. The phylogenetic analyses of HA, NA and M genes showed that A/duck/Chongqing/BB/2012 and A/chicken/Chongqing/BN/2012 were distant relatives. However, both of the two strains have the same HA gene proteolytic cleavage sites, RERRR-KR, which accorded with the molecular characteristic of high pathogenic AIV. These results could provide more information for the analysis of the sequence of avian influenza virus H5N1, including variation analysis, evolutionary analysis, as well as the development of vaccine.Key words: Avian influenza virus; H5N1 subtype; gene clone; sequence analysis; phylogenic analysis
2016-10-19
重庆市科技攻关计划项目(cstc2012gg-yyjs80014);重庆市科技人才培养计划项目(cstc2013kjrc-1jrcpy80001)
胡仁建(1968—),女,重庆人,博士研究生,高级实验师,主要从事微生物与生化药学研究,E-mail:hrj@cqut.edu.cn。
胡仁建, 顾盼, 蔡家利, 等.重庆两株H5N1流感病毒的HA、NA基因克隆及序列分析[J].重庆理工大学学报(自然科学),2017(1):67-74.
format:HU Ren-jian, GU Pan, CAI Jia-li, LIU Hai-jun,et al.HA and NA Gene Clone and Sequence Analysis of Two H5N1 Virus Isolates from Chongqing[J].Journal of Chongqing University of Technology(Natural Science),2017(1):67-74.
10.3969/j.issn.1674-8425(z).2017.01.011
S855.3
A
1674-8425(2017)01-0067-08
猜你喜欢
当代党员(2022年9期)2022-05-20
当代党员(2022年9期)2022-05-20
科学大观园(2022年2期)2022-01-23
西藏艺术研究(2021年4期)2021-06-02
重庆与世界(2019年12期)2019-09-10
农产品市场周刊(2019年8期)2019-06-30
重庆社会科学(2017年12期)2017-02-06
红岩春秋(2016年8期)2016-05-14
动物医学进展(2015年10期)2015-12-07
特产研究(2014年4期)2014-04-10
