
合成免疫学①
2017-02-27 05:56李桉棋
中国免疫学杂志 2017年2期
曹 玲 李桉棋 李 峰 黄 波 张 毅
(郑州大学第一附属医院生物细胞治疗中心,郑州450052)
合成免疫学①
曹 玲 李桉棋 李 峰 黄 波②张 毅
(郑州大学第一附属医院生物细胞治疗中心,郑州450052)
合成免疫学是合成生物学和免疫学的综合。合成免疫学是一门新兴的科学领域,它是免疫学和合成生物学的交叉学科,目的是利用生物技术手段来合理调节及控制免疫反应从而使患者获益(图1)。 在出现免疫系统紊乱时需要校正免疫反应,例如,在风湿性关节炎(RA)中应用免疫细胞来抑制促炎介质。同时也可以利用免疫细胞来治疗疾病,例如,利用免疫细胞的细胞毒作用来清除肿瘤。干扰免疫系统可以通过使用重组细胞因子或重新编程免疫细胞来实现,重组细胞因子和生长因子或治疗性抗体已经应用于患者,但更多的基于合成免疫学原理的治疗药物和治疗手段尚处于临床前测试或临床研究阶段。在分子水平上,研究人员通过改造和修饰抗体衍生物和抗体模拟物等新的治疗药物以提高其治疗的安全性和有效性。在细胞水平上,人们则通过工程化免疫细胞改变其效应目标、从而提高免疫细胞的功能。合成免疫学一种可以合理设计并构建复杂免疫反应功能的合成系统,可以最大限度的实现调控免疫反应的目的,它主要通过人工合成的成分来控制免疫反应,如纳米材料、免疫佐剂和疫苗等。这些技术目前仍处于探索阶段,为化学家、生物学家和临床医生带来了巨大的挑战。
在本综述中,我们讨论了免疫系统调控的方法和途径以及带来的挑战,并且展望了未来的治疗策略。我们认为,合成免疫学的应用必将成为一种越来越强大的治疗途径。
1 免疫系统的分子工程化
多种治疗性分子可以用来干扰免疫系统,在这篇综述中,我们仅讨论基于人源成分的工程化分子,例如治疗性抗体、嵌合蛋白、抗体衍生物和抗体类似物。第一代内源工程化分子包括重组人细胞因子和生长因子。
1.1 治疗性抗体 20世纪中期,研究人员开始利用治疗性抗体治疗人类疾病。20世纪70年代,Milstein 和Kohler报道了第一个工程化的单克隆抗体,莫罗单抗CD3(OKT3),它通过干扰T细胞的激活应用于移植排斥,在1986年被美国食品和药品监督管理局(FDA)批准上市。目前,约30种治疗性抗体已被批准,并且超过300种化合物正在进行临床试验[1]。这些治疗性抗体可在体外合成(人抗体),或者通过小鼠产生,然后被工程化以生产更多的模拟人源分子(嵌合和人源化抗体)。
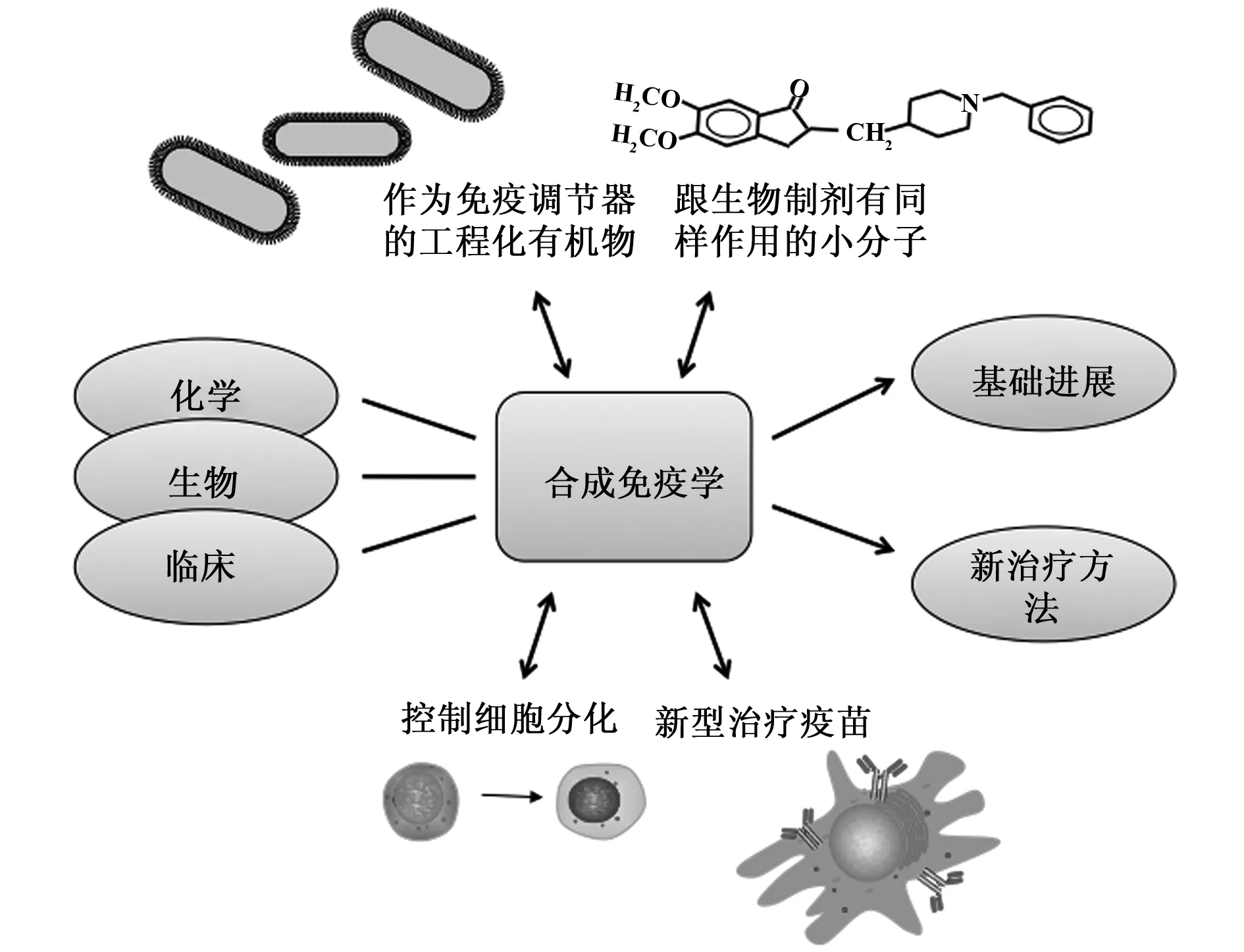
图1 合成免疫学结构图Fig.1 Diagram of synthetic immunologyNote: Chemistry,biology and clinical disciplines were combined together,various treatment approaches such as synthetic molecules,cells or other products were applied to promote the development of synthetic immunology
抗体的主要优点是其突出的亲和力和特异性以及较长的血浆半衰期(表1)。此外,抗体与配体结合时可以通过多种机制起作用,第一,抗体的结合可以中和配体的功能。第二,抗体可以被改造从而向靶向位置递送有毒物质。第三,抗体可介导ADCC效应(抗体依赖性细胞介导的细胞毒性)。第四,抗体与细胞的结合可介导CDC效应(补体依赖性细胞毒性),通过补体因子诱导凋亡。
然而,使用治疗性抗体也有局限性。因其体积较大和空间构象的限制,治疗性抗体不能穿过细胞膜,只能与细胞外的分子相互作用,而且组织渗透性较低。相对较高的价格也是限制治疗性抗体广泛应用的原因。
下面我们将讨论几种干扰人类免疫系统的抗体,包括针对免疫细胞上促炎性分子或表面受体的抗体以及可以介导免疫细胞效应功能的抗体。
1.1.1 单克隆抗体 单克隆抗体从非人类物种制备,并且不需人源化便可以直接使用。例如,OKT3是一种针对CD3的单克隆抗体,已经被用于阻断器官移植后T细胞的活化。新一代单克隆抗体已实现可以双重特异性地与目标相互作用,例如,卡妥索单抗是三功能性抗体,可以双特异性地靶向过度表达在多种实体癌的上皮细胞黏附分子(EpCAM)的两个跨膜糖蛋白,同时可以结合T细胞特异性表面蛋白CD3,因而卡妥索单抗可借助其Fc区吸引免疫细胞,同时可以通过抗CD3抗体招募T细胞和ADCC[2]。这种由小鼠IgG2a,大鼠IgG2b杂交产生的抗体在2009年被批准用于EpCAM阳性肿瘤恶性腹水的治疗。
治疗性抗体的安全性不仅仅是免疫原性的问题;非人源抗体成分形成的人抗药物抗体带来的风险尤其值得重视。研究发现,在卡妥索单抗药代动力学第Ⅱ阶段研究中,接受治疗的所有患者体内都可以检测到抗药物抗体[3]。
1.1.2 嵌合单克隆抗体 嵌合单克隆抗体,是由小鼠可变区与人恒定区结合,形成含有约75%人源序列的蛋白[4]。与非人源的单克隆抗体相比,使用人源的恒定区序列作为铰链区能够减少人体抗药抗体的形成。
几种治疗性抗体(Ximab)已成功进入临床应用。靶向抗肿瘤坏死因子(TNF)的嵌合单克隆抗体英利昔单抗(Infliximab)用于治疗几种严重型的自身免疫性疾病,包括类风湿性关节炎和炎症性肠疾病。 利妥昔单抗(Rituximab),抗CD20嵌合人鼠单抗,不仅用于治疗B细胞恶性肿瘤,而且还可用于治疗常规疗法治疗失败的自身免疫疾病,如类风湿性关节炎和抗中性粒细胞胞浆抗体(ANCA)相关小血管炎。机制是抗体结合CD20阳性B细胞,引起ADCC,CDC,进而激活CD20导致B细胞凋亡耗尽[5]。巴利昔单抗(Basiliximab),一种针对T细胞表面白细胞介素2受体α链的抗体,通过阻断T细胞活化从而防止器官移植的排斥反应。
1.1.3 人源化单克隆抗体 一种新的治疗性抗体,由鼠源的抗原结合序列(又称为互补决定区)与人类抗体铰链区连接构成。非人源的序列减少到抗体序列的5%,进一步降低了免疫原性[4]。其中,阿仑单抗(Alemtuzumab)可以结合淋巴细胞表面CD52分子,通过ADCC作用清除这些细胞。另一种人源化单克隆抗体,那他珠单抗(Natalizumab),可以结合整合素α4(α4 integrin),从而阻断淋巴细胞的迁移,用于多发性硬化和克罗恩病的治疗[6]。尽管这种抗体的免疫原性已经明显降低,但可以激活一些高致敏反应[7],有证据表明那他珠单抗会增加感染的风险,治疗过程中并发进展性多病灶性脑白质病。具体说来,因为免疫细胞迁移受限,在中枢神经的免疫监视系统减少,从而使病毒变得活跃。
表1 基于合成免疫学的治疗手段
Tab.1 Therapies based on synthetic immunology

方法疾病种类优势劣势重组蛋白免疫缺陷、自身免疫疾病经济、安全、低免疫原性、良好的组织渗透性体内半衰期短合成蛋白自身免疫疾病高亲和性和特异性有产生免疫原性的危险,体内半衰期短,价格高人源性抗体自身免疫疾病高亲和性和特异性、ADCC、CDC、携带毒性产物价格高,组织渗透性低双特异性单克隆抗体肿瘤免疫治疗高亲和性和特异性、双特异性、ADCC、CDC、携带毒性产物有产生免疫原性的危险,体内半衰期短,价格高抗体衍生物肿瘤免疫治疗高亲和性和特异性、良好组织渗透性、比抗体便宜体内半衰期短抗体模拟物自身免疫疾病经济、高亲和性和特异性体内半衰期短同源造血干细胞基因治疗免疫缺陷治疗疾病有恶变的危险过继性免疫治疗肿瘤免疫治疗效能好、持久性治疗价格高、肿瘤微环境的免疫抑制性导致的低效率
1.1.4 人单克隆抗体 近年来研究比较多的人单克隆抗体全部为人源序列,是由表达人源免疫球蛋白或杂交瘤的噬菌体、转基因小鼠制备而成[8]。例如来自噬菌体并靶向肿瘤坏死因子(TNF)的阿达木单抗,以及FDA批准应用于黑色素瘤的Ipilimumab 和Nivolumab两种人单克隆抗体。
免疫治疗最大的挑战是要克服肿瘤部位的免疫抑制微环境。Ipilimumab 和Nivolumab两种抗体通过激活T细胞来对抗恶性肿瘤,在 免疫治疗领域取得了重大突破。当CTLA-4与抗原提呈细胞表面的B7结合时会导致T细胞无能,而Ipilimumab通过与T细胞表面共刺激分子CTLA-4结合而阻断两者的结合,从而影响了CTLA-4的抑制作用(图2)。用Ipilimumab治疗恶性黑色素瘤可以延缓肿瘤生长,延长病人平均生存期9到12个月[9]。Nivolumab 是一种人源IgG4 抗体,通过靶向T细胞表面程序性死亡分子-1(PD-1)促进T细胞的激活(图2),从而延长恶性黑色素瘤病人的生存期[10,11]。 研究发现,PD-1单抗免疫治疗通过修复T细胞免疫反应而达到清除肿瘤的目的[12]。两种T细胞调节剂Ipilimumab 和Nivolumab在临床上的联合应用可快速清除恶性黑色素瘤病人的肿瘤,并延长他们的生存期,并且副作用未增加,仍在可耐受的范围[13]。Nivolumab是第一个靶向PD-1的单克隆抗体且对无法手术或者转移性黑色素瘤、非小细胞肺癌(Non-small cell lung cancer,NSCLC)、转移性肾癌都有良好的临床疗效[10,14]。Nivolumab的临床实验显示,在黑色素瘤入组患者中可达到30% 到40%的持续客观反应率(Objective response rates,ORR)(NCT00730639,NCT01721772,NCT01844505)[15,16]。
另一种PD-1通路上的单克隆抗体 anti-PD-L1 BMS-936559 (MDX-1105)已经在临床一期试验中显示出良好的疗效(NCT00729664) (图2)[17]。有文献报道,anti-CTLA4与anti-PD-1相比,临床疗效较差[18],原因可能在于anti-CTLA4诱导的是T细胞对肿瘤抗原的非特异性识别[19-24]。因此,临床应用时应该优先考虑使用靶向PD-1的单克隆抗体[25]。
临床中常使用促炎性细胞因子来增强对肿瘤细胞的免疫反应。多种细胞因子已经被批准用于治疗恶性肿瘤。由于这些细胞因子是全身用药,并且这些细胞因子代谢比较慢,所以经常导致严重的不良反应。因此,可将细胞因子融合到靶向肿瘤的抗体中,在提高癌症免疫疗法疗效的同时也可以降低不良反应发生率[26]。一些融合了Fc结构域或抗体不同区域的细胞因子,已经进入肿瘤免疫治疗的Ⅰ、Ⅱ期临床试验[27]。
正在临床研究测试的治疗性单克隆抗体逐年在增加,许多公司正在生产靶向促炎性细胞因子或免疫细胞表面受体的单克隆抗体来治疗自身免疫性疾病。但这些抗体靶向的细胞因子和免疫细胞是机体对病原体产生炎症反应所必需的,在使用这些抗体时虽然能够治疗自身免疫病,却抑制了机体对病原体的免疫应答,因此容易合并感染[28]。总之,单克隆抗体有着出色的特异性和亲和性,但与传统免疫制剂相比不良反应并没有减少。
1.2 嵌合蛋白 调节免疫系统的主要机制是结合或者中和促炎细胞因子,因此我们可以使用治疗性抗体,也可以设计出能够结合炎性细胞因子的嵌合蛋白质 。当工程化的嵌合蛋白与靶向的细胞因子结合后,因嵌合蛋白中主要为免疫球蛋白(图3A)或白蛋白发挥作用,因此具有较高的亲和性和特异性。
例如,为了阻断炎症因子TNF的作用,研究者将TNF受体2(TNFR2)与人源IgG1的Fc区相连设计出了嵌合蛋白依那西普(Etanercept),目前已被广泛应用于临床治疗自身免疫性疾病(图3B)。

图2 用于免疫检查点阻断的单克隆抗体Fig.2 Human monoclonal antibodies for immuno-check-point blockade
1.3 抗体衍生物 抗体衍生物是一种新的抗体样分子,从免疫球蛋白的可变区分离得到的抗体衍生物,具有生产成本低、组织穿透性强、免疫原性较低[29]、血清半衰期较短等优点,目前已在免疫紊乱疾病及恶性肿瘤中开展临床试验。抗体衍生物通过接头序列可以连接多个可变结构域,因此可以包含几个独立的抗原结合位点,在体内可结合多个化合物,目前多用于招募毒性免疫细胞进入肿瘤组织并靠近靶细胞。
1.3.1 双特异性T细胞结合(BiTE)抗体 BiTE抗体是一种由可变结构域的重链和轻链组成的单链多肽,重链和轻链通过柔性接头序列连接[30](图4A),可以将抗CD3可变结构域连接到任何靶点的可变区。因此,BiTE 抗体可以招募细胞毒性T细胞直接攻击目标靶点。
1.3.2 双亲和力重定向(DART)技术 像BiTE一样,DART抗体可以结合两个目标。然而,DARTs与BiTE在结构上不同:DARTs包含两个多肽链,重链针对一个目标,轻链针对另一个目标,重链和轻链通过链间二硫桥相连(VLa-VHb或VLb-VHa)(图4B)[31]。
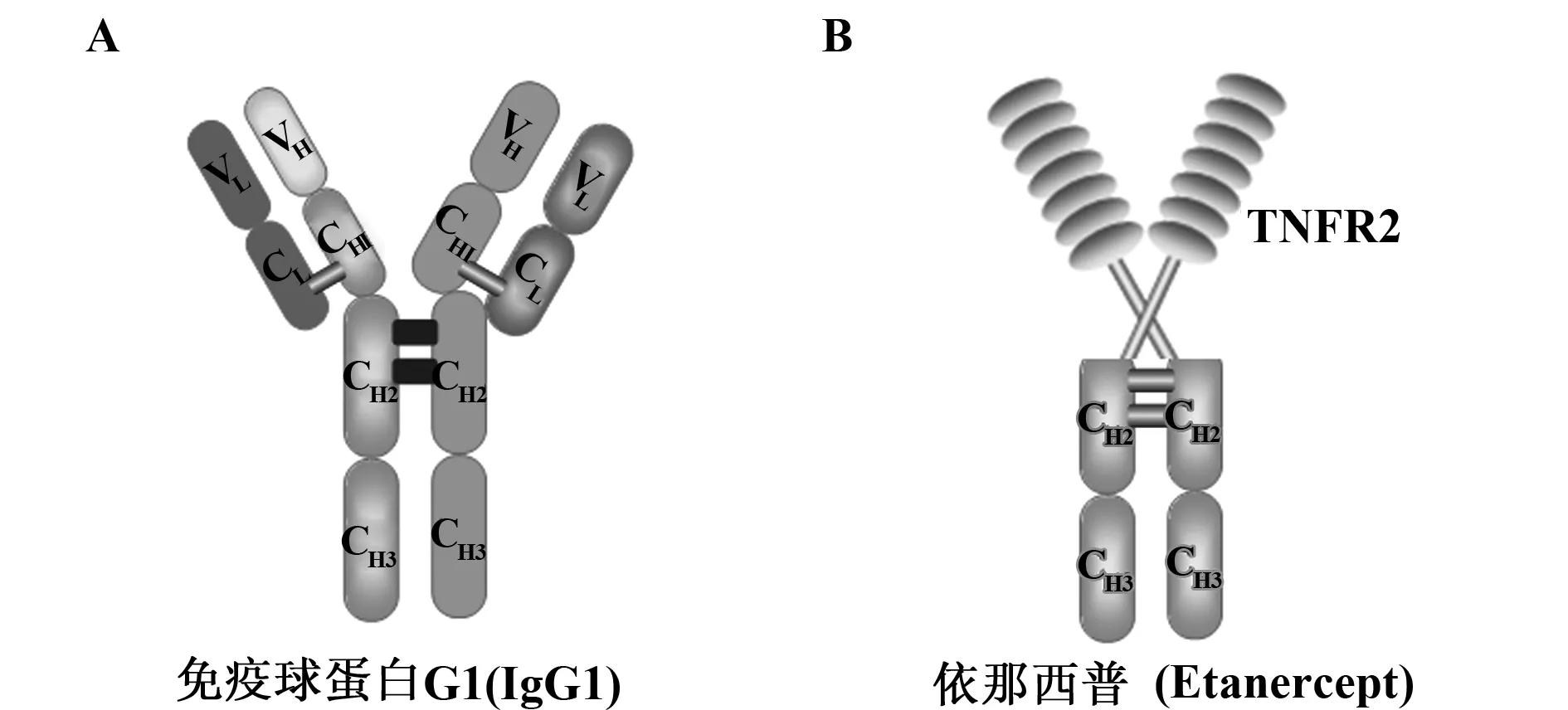
图3 嵌合蛋白Fig.3 Chimeric proteinsNote: A,bispecific IgG1;B,Synthetic protein etanercept is chimeric molecules linking IgG1 to target TNF-binding protein TNFR2.
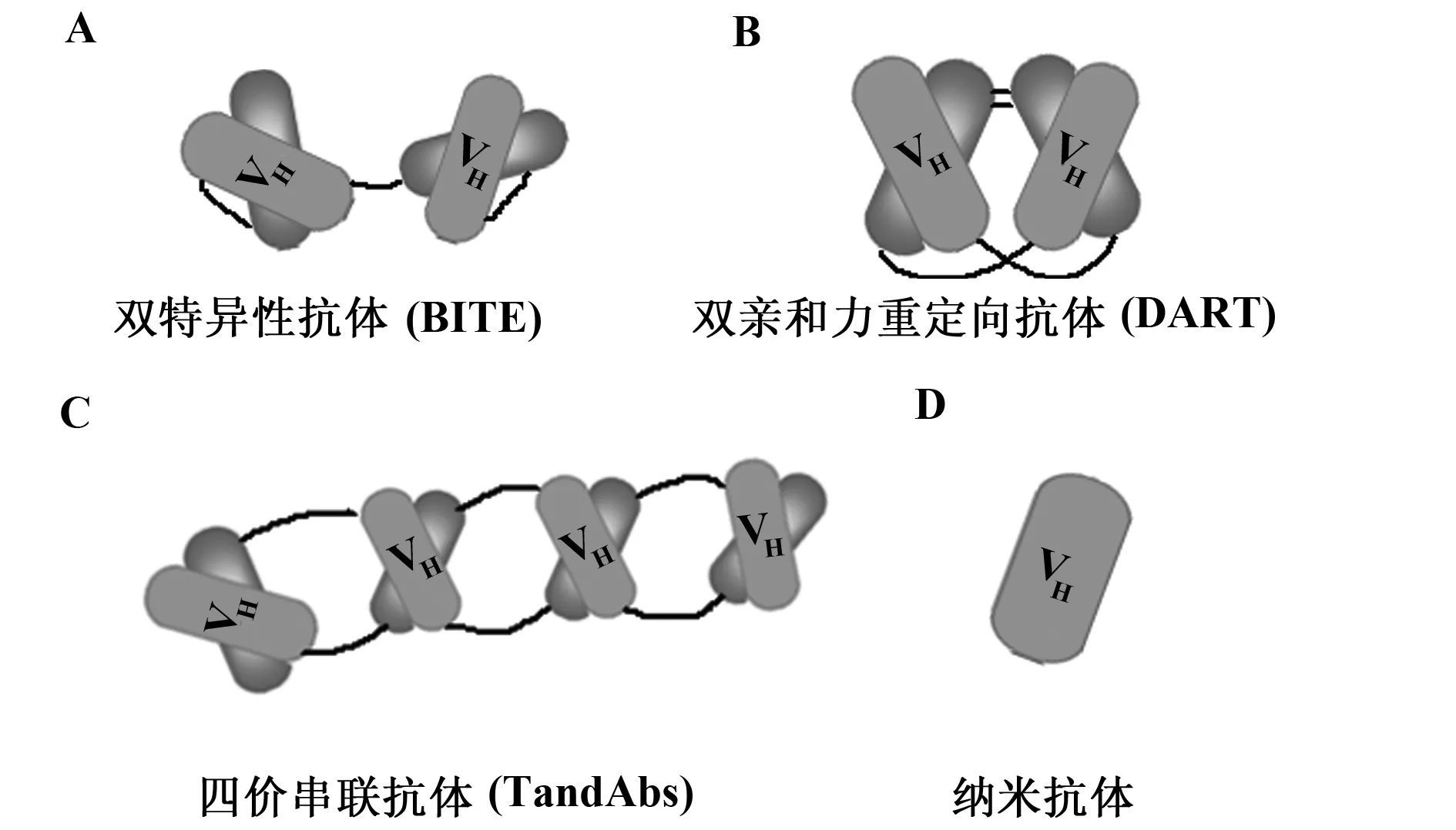
图4 抗体衍生物 Fig.4 Antibody derivatives
1.3.3 四价串联抗体(TandAbs) TandAbs包括两个二聚化的多肽链和四个抗原结合位点(图4C),分子量为110 kD(VLa-VHb-连接体 -VLb-VHa)[31]。
1.3.4 纳米抗体 纳米抗体是从由重链组成的单域抗体,这些分子包括抗体重链(VHH)可变域(图4D),分子量仅为12~15 kD。多种人源化和序列优化的纳米抗体已进入临床药物研究阶段。目前已有3个纳米抗体正在自身免疫性疾病的治疗中进行功能评估[32],有报道指出,2%接受纳米抗体治疗的受试者出现了抗药物抗体,但是这些抗体并没有影响疗效。
目前已经出现纳米抗体工程结合促炎细胞因子的研究,例如 Ozoralizumab,它是一种双特异性抗体衍生物,由3个 VHH组成,有两个TNF的结合位点。此纳米抗体在RA的治疗中进行临床试验[33]。有效性和安全性试验将进一步证实纳米抗体是否优于治疗性单克隆抗体。
多特异性抗体和抗体衍生物使得效应细胞向靶细胞的迁移不依赖于ADCC效应,这将为更多的干扰免疫反应的方法提供研究基础。主要的问题还是在于找到合适的肿瘤抗原作为有效的治疗靶点。
1.4 抗体模拟物 抗体模拟物是包含了抗原结合位点的合成蛋白,这些分子包含的单个多肽序列来源于现有的人源支架蛋白。抗体模拟物也是高效的蛋白质,分子量低但可提供高特异性。在过去的20年已经发现约50个不同的蛋白支架[31],其中一些已经用于过敏性疾病和自身免疫性疾病的临床前和临床研究。
1.4.1 设计的锚蛋白重复蛋白(DARPins) 3~5个完整设计的锚蛋白重复支架决定了DARPins的结合特异性,这些中央基序侧翼的N、C端帽基序遮挡了疏水区域,从而增加了DARPins的溶解度(图5A)。 DARPins 缺乏T 细胞表位, 因此其免疫原性较低,并且在临床试验中已观察到这种现象。
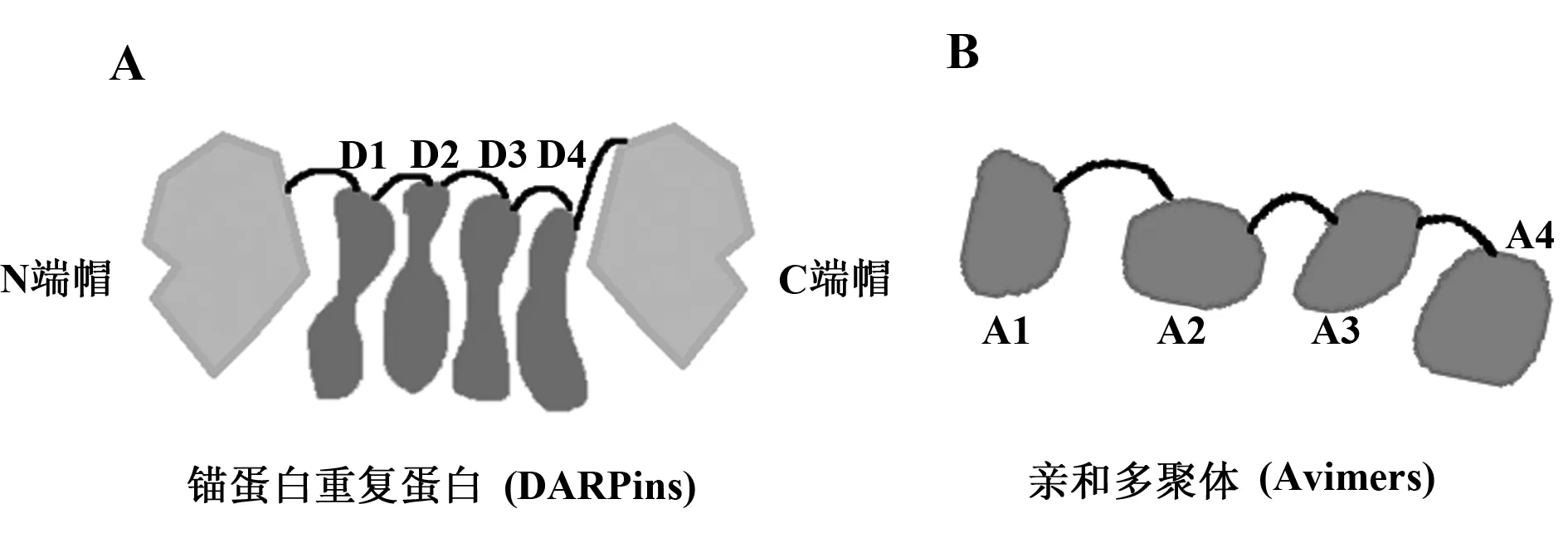
图5 抗体模拟物Fig.5 Antibody mimeics
1.4.2 亲和多聚体(Avimers) Avimer的结构是基于所谓的A区,A区主要存在于各种细胞表面受体的胞外部分,它以低亲和力与同一靶点的不同表位相结合(图5B)。A结构域的亲和力可以通过单一域多聚化而增加,这是由遗传的低聚化来实现的。事实上,组合A结构域已由外显子洗牌产生[34]。目前研究最多的Avimer,称为AMG220,是一个12 kD的脚手架蛋白,其中包含低密度脂蛋白受体的三个A域,可以高亲和力结合IL-6,这个Avimer正在克罗恩病中进行Ⅰ期临床试验[35]。
2 免疫细胞的工程化
除了合成蛋白调控免疫系统之外,大量研究关注于利用基因工程手段体外改造免疫细胞。因此在本部分中我们将讨论免疫细胞工程化及其临床应用。合成免疫学也包括调节其他细胞和机体来干扰人体免疫系统,如间充质干细胞(MSC)治疗、工程化细菌用于肠道慢性炎症、白介素-2(IL-2)、HIV融合抑制肽等[36-39]。
免疫细胞工程化目前已经应用于在原发免疫缺陷患者中对功能缺陷的免疫细胞进行体外重组,在获得性免疫缺陷患者中通过造血干细胞(HSC)基因治疗来抑制HIV感染,或者利用免疫细胞的特性通过过继性免疫治疗来对抗肿瘤。工程化自体免疫细胞疗效持久,可以达到治愈疾病的目的。
2.1 细胞表面工程化——在细胞膜上添加成分 现今有多种改造策略将某些分子或化合物负载至免疫细胞表面可以调节效应细胞的功能。自分泌因子可以用脂质纳米颗粒包装,通过共价键连接到细胞表面,从而实现低浓度介质持续刺激细胞的目的。细胞表面工程改造还包括通过细胞因子介导体内T细胞的大量扩增,从而增强T细胞的杀伤作用[40]。
2.2 自体HSC基因治疗——治疗免疫缺陷 在自体HSC基因治疗中,对基因缺陷的细胞群通过基因添加、修改、敲除等来恢复这些基因的表达,从而缓解患者的临床症状,使病情不再进展,甚至可能达到治愈。当前,免疫缺陷的主要治疗方法是通过负载野生型基因序列的载体,以及通过基因修正或者基因敲除来治疗HIV感染患者。
2.2.1 基因添加 在3种HSCs治疗的方法中,目前临床试验及临床前应用最多的是基因添加,第一个成功的案例发生在2000年,一个患有X染色体遗传的严重免疫缺陷的儿童患者通过自体骨髓移植恢复了免疫系统的功能,移植的骨髓细胞中带有小鼠白血病载体负载的野生型IL-2Rα受体[41]。
2.2.2 基因修正 由于基因修正能在不影响表达调控的前提下改变基因,所以近来受到了极大的关注。利用这种方法,可以使用锌指结构核酸酶,归巢内切酶,转录激活因子来纠正基因突变或制造新的突变基因。由于采用了不同的修复手段,结果可能造成基因的破坏,重构或剪切。
例如,在CCR5中引入突变,使得T细胞能抵抗HIV的感染[42]。将人类HSCs通过锌指结构核酸酶技术设计突变转入到CCR5,将这些细胞移植到免疫功能不全的小鼠后,接种HIV病毒后,病毒滴度明显低于未突变小鼠[43]。HIV感染者使用自体转导的锌指结构核酸酶CD4+T细胞的初步研究刚刚完成,研究显示这种方法是安全的[44]。
2.2.3 基因敲除 沉默单基因及复杂基因网络的技术逐渐应用于基因治疗。主要手段是利用发卡RNA(shRNAs)来沉默目的基因。目前,沉默CCR5的免疫细胞正在艾滋病患者中进行临床试验[45]。其策略是利用自体转导带有编码CCR5-shRNA慢病毒载体的CD4+T细胞和CD34+造血干细胞,同时联合HIV抑制剂来治疗HIV患者。Ⅰ期临床试验的评估目标首先是安全性和可行性,然后是CD4+T细胞剂量以及病毒载量。
2.3 过继免疫治疗 现有的过继性免疫治疗主要用于肿瘤细胞的清除,具体来说,从患者体内分离出免疫细胞,在体外进行扩增和基因修饰使得它们重定位自己的效应功能,随后回输至患者(图6),基因工程化的效应细胞将识别它们的新靶点并沉默或者杀死肿瘤。
尽管过去的10年已经在临床试验中应用了不同的治疗策略,但工程化免疫细胞过继性免疫治疗到目前为止仅显示了温和的效应。CAR(嵌合抗原受体)修饰T细胞在白血病和淋巴瘤中显示了鼓舞人心的疗效,但在靶向实体瘤时仍有局限性。主要问题是要克服实体瘤肿瘤部位的免疫抑制微环境,同时要实现回输至宿主体内后有足够的持久性[46]。目前临床试验中应用的CAR只有单克隆特异性,已有研究发现由于发生了抗原逃逸而导致CAR-T无法发挥长期的抗肿瘤效应[47]。此外,这种技术成本比较高,而且有可能发生致命的不良反应[48,49]。
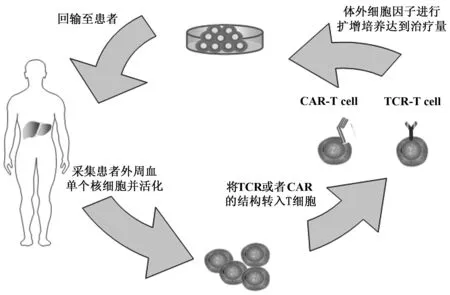
图6 基因工程化T细胞的制备及回输流程Fig.6 Preparation and infusion process of gene-modified T cells
与单克隆抗体免疫制剂相比,过继性免疫治疗的主要优点是利用了免疫细胞自身的抗肿瘤免疫效应,单次回输后即可主动迁移至肿瘤部位,发生抗原特异性扩增后在一定时间内可以持续存在。
2.3.1 工程化T细胞 研究人员可以使用多种技术对T细胞进行基因修饰,使其表达高亲和力TCRs(T细胞受体),从而获得对靶抗原的特异性。研究人员最常使用逆转录病毒来介导T细胞的基因修饰,转座子系统和mRNA电转染技术同样可以成功将遗传物质转至T细胞。现有临床试验使用的TCRs主要有以下几个来源:一是从对过继细胞治疗有反应的患者中分离得到编码TCR的DNA序列,这种技术可以从肿瘤微环境中分离出自体T细胞,然后在体外进行扩增,得到的TCR序列随后被克隆进逆转录病毒载体并转染至HLA匹配患者的T细胞上。第二,TCR的遗传信息可以从人源化小鼠中(表达MHCⅠ/Ⅱ分子并可以识别肿瘤抗原)分离得到。表达小鼠TCRs的人源工程化T细胞已被成功用于靶向肿瘤抗原CEA和PMEL蛋白。第三,利用测序技术鉴定出肿瘤特异性突变,然后体外合成这些突变基因或者肽并表达于患者自体抗原提呈细胞(APC),然后将自体T细胞或者健康供者来源的T细胞与这些表达了患者自身肿瘤突变的APC共培养,将获得能够识别患者自身所有突变的TCR-T细胞[50,51]。第四,设计并构建CAR的结构并通过病毒载体或其他方法转染至患者来源的T细胞中。这种情况下,抗体的单链可变区(scFv)被嵌合至TCR的跨膜区和胞内区,下游串联了可以激活T细胞的共刺激分子。因此,CARs可以识别靶细胞表面非MHC限制性抗原,而TCRs主要识别被APC(抗原提呈细胞)处理后以肽的形式与MHC分子形成复合物的胞内抗原。
靶向CD19阳性白血病和淋巴瘤的CAR-T细胞在临床试验中已经取得了良好的疗效,首批4例患者的病情得到了完全缓解[52,53],但是在另外一例使用同样CAR-T细胞治疗的患者中出现了CD19阴性白血病的复发[54]近两年来,CAR-T细胞免疫治疗在B细胞恶性肿瘤中取得了很好的疗效。Maude等[55]报道了30例急性淋巴细胞白血病(ALL)患者入组靶向CD19的CAR-T细胞(CTL019)治疗的临床试验结果,完全缓解率达到了90%,最长生存时间超过了2年,而且入组患者均为多次复发或者对两个或更多再次诱导方案无效者,其中60%患者为自体干细胞移植后复发,这样的疗效无疑非常振奋人心。Porter等[56]报道了14例复发或难治慢性淋巴细胞白血病患者CAR-T治疗的效果,虽然总的缓解率仅为56%,但观察到了长达53个月的无病生存期,这样显著的疗效用传统疗法基本是不可能实现的。TCR-T细胞免疫治疗也在临床试验中显示了可观的疗效,Rapoport等[57]报道的NY-ESO-1特异性TCR-T细胞在治疗20例多发性骨髓瘤患者时,临床有效率高达80%(16/20),完全缓解率(CR)也达到了70%(14/20),而且中位无疾病进展生存期(PFS)长达19.1个月。
尽管如此,Zhang等[58,59]对近年来CAR-T细胞免疫治疗在B细胞恶性肿瘤中的临床试验进行Meta分析后发现,CAR-T细胞免疫治疗可将ALL患者总的缓解率提高至90%,而在CLL(慢性淋巴细胞白血病)中仅提高至42%,并指出ALL患者效果优于CLL可能源于CLL患者的体内免疫抑制更强,因此有必要联合其他治疗以提高CAR-T的疗效。
近年来研究人员开发出多种方法与CAR联合,来增加CAR-T免疫治疗的疗效和安全性。有研究发现,PD-1阻断联合CAR-T治疗可以明显延长CAR-T细胞在实体瘤中的存活时间并增强抑制PD-L1+肿瘤细胞的能力并在转移性黑色素瘤患者中增加CAR-T细胞的持久性从而提高CAR-T细胞的抗肿瘤效应[60],或者采用基因工程手段将PD-1构建进CAR-T细胞中从而增强CAR-T细胞在肿瘤部位的浸润[61]。一种提高CAR-T细胞特异性的方法是设计出仅能靶向和杀伤表达两种抗原靶细胞(例如肿瘤细胞)的CAR,这种新型的CAR不会识别表达一种或者不表达抗原的细胞(例如正常组织细胞),因此提高了特异性和治疗的安全性[62]。此外,还出现了将抗体scFv区与免疫抑制受体CTLA-4或PD-1嵌合的抑制性CARs[63]。研究人员还利用基因工程使T细胞异位表达T细胞刺激因子IL-12,IL-12/CAR的组成性内源分泌在没有化疗预处理的情况下即可引起表达靶向抗原的肿瘤细胞的凋亡,并且可以抵抗Treg细胞介导的免疫抑制[64]。最近发表在Science上的一篇报道更是提出了通过一个小分子开关来控制CAR-T功能的新方法[65]。这些新的合成免疫学的产物在优化T细胞功能及减少不良反应方面有重要的意义。
2.3.2 应用经过工程改造的NK细胞(自然杀伤细胞)根除肿瘤 自体或异体NK 细胞已广泛用于多种恶性肿瘤治疗的临床试验中[66]。NK 细胞最早在血液中发现,回输NK细胞的免疫疗法在恶性血液系统肿瘤中已经获得了成功[67]。首先,NK 细胞被分离出来(主要来自外周血单核细胞),然后,在体外用细胞因子刺激并扩增至治疗剂量后回输至患者体内。尽管临床试验数据表明自体NK细胞移植是安全的,但是临床效率却差强人意[66]。相反,异体NK细胞在肿瘤治疗中相对比较成功,包括转移性恶性黑色素瘤、肾癌、霍奇金淋巴瘤和急性髓系白血病[68]。
基因工程改造的NK细胞和NK细胞系的抗肿瘤效果已在临床前研究中被广泛证实,多项研究表明,联合细胞因子 (IL-2、IL-15、SCF)基因修饰NK细胞和用CAR (CD19、CD20、ErbB2、CD33、CEA、GD2)来定向NK靶细胞的新技术能够提高NK细胞的杀伤功能、增强NK细胞的扩增能力、生存能力及其靶向特异性,从而增强NK细胞的抗肿瘤效应[66]。这些新技术在临床试验中能否成功有待更多临床试验来验证。
3 未来治疗策略
基于合成免疫学的理念,近年来出现并上市了多种新的分子水平上的多特异性抗体、抗体衍生物和抗体模拟物,而且可以通过重新编程免疫效应细胞来改变效应细胞的功能。目前,过继免疫疗法主要应用于肿瘤领域,目的是通过基因工程手段增强T细胞的抗肿瘤能力。现有免疫治疗存在的主要问题就是免疫应答的特异性较低从而有可能导致脱靶效应,例如,靶向CD19/CD20来杀伤白血病细胞在清除肿瘤细胞的同时也会大幅度的抑制正常B细胞功能从而干扰人体正常免疫系统。在不久的将来,研究者必定会更多关注于蛋白复合体如何与特异性更强的细胞亚群联合应用,以及如何通过CAR技术来整合多重特异性。在未来,工程化分子和工程化细胞在肿瘤相关领域将开展更大规模的研究,在其他领域也必将有越来越广泛的应用。
致谢:衷心感谢中国工程院杨胜利院士在本文书写过程中给予的指导和帮助。
[1] McEnaney PJ,Parker CG,Zhang AX,etal.Antibody-recruiting molecules:an emerging paradigm for engaging immune function in treating human disease[J].ACS Chem Biol,2012,7(7):1139-1151.
[2] Chelius D,Ruf P,Gruber P,etal.Structural and functional characterization of the trifunctional antibody catumaxomab[J].MAbs,2010,2(3):309-319.
[3] Ruf P,Kluge M,Jager M,etal.Pharmacokinetics,immunogenicity and bioactivity of the therapeutic antibody catumaxomab intraperitoneally administered to cancer patients[J].Br J Clin Pharmacol,2010,69(6):617-625.
[4] Clark M.Antibody humanization:a case of the ′Emperor′s new clothes′?[J].Immunol Today,2000,21(8):397-402.
[5] Amoroso A,Hafsi S,Militello L,etal.Understanding rituximab function and resistance:implications for tailored therapy[J].Front Biosci (Landmark Ed),2011,16:770-782.
[6] Rudick R,Polman C,Clifford D,etal.Natalizumab:bench to bedside and beyond[J].JAMA Neurol,2013,70(2):172-182.
[7] Hansel TT,Kropshofer H,Singer T,etal.The safety and side effects of monoclonal antibodies[J].Nat Rev Drug Discov,2010,9(4):325-338.
[8] Vincent KJ,Zurini M.Current strategies in antibody engineering:Fc engineering and pH-dependent antigen binding,bispecific antibodies and antibody drug conjugates[J].Biotechnol J,2012,7(12):1444-1450.
[9] Wolchok JD,Hodi FS,Weber JS,etal.Development of ipilimumab:a novel immunotherapeutic approach for the treatment of advanced melanoma[J].Ann NY Acad Sci,2013,1291:1-13.
[10] Topalian SL,Hodi FS,Brahmer JR,etal.Safety,activity,and immune correlates of anti-PD-1 antibody in cancer[J].N Engl J Med,2012,366(26):2443-2454.
[11] Ott PA,Hodi FS,Robert C.CTLA-4 and PD-1/PD-L1 blockade:new immunotherapeutic modalities with durable clinical benefit in melanoma patients[J].Clin Cancer Res,2013,19(19):5300-5309.
[12] Sznol M,Chen L.Antagonist antibodies to PD-1 and B7-H1 (PD-L1) in the treatment of advanced human cancer[J].Clin Cancer Res,2013,19(5):1021-1034.
[13] Wolchok JD,Kluger H,Callahan MK,etal.Nivolumab plus ipilimumab in advanced melanoma[J].N Engl J Med,2013,369(2):122-133.
[14] Brahmer JR,Drake CG,Wollner I,etal.Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors:safety,clinical activity,pharmacodynamics,and immunologic correlates[J].J Clin Oncol,2010,28(19):3167-3175.
[15] Topalian SL,Sznol M,McDermott DF,etal.Survival,durable tumor remission,and long-term safety in patients with advanced melanoma receiving nivolumab[J].J Clin Oncol,2014,32(10):1020-1030.
[16] Robert C,Long GV,Brady B,etal.Nivolumab in previously untreated melanoma without BRAF mutation[J].N Engl J Med,2015,372(4):320-330.
[17] Brahmer JR,Tykodi SS,Chow LQ,etal.Safety and activity of anti-PD-L1 antibody in patients with advanced cancer[J].N Engl J Med,2012,366(26):2455-2465.
[18] Robert C,Schachter J,Long GV,etal.Pembrolizumab versus Ipilimumab in Advanced Melanoma[J].N Engl J Med,2015,372(26):2521-2532.
[19] Karandikar NJ,Vanderlugt CL,Walunas TL,etal.CTLA-4:a negative regulator of autoimmune disease[J].J Exp Med,1996,184(2):783-788.
[20] Perrin PJ,Maldonado JH,Davis TA,etal.CTLA-4 blockade enhances clinical disease and cytokine production during experimental allergic encephalomyelitis[J].J Immunol,1996,157(4):1333-1336.
[21] Luhder F,Hoglund P,Allison JP,etal.Cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) regulates the unfolding of autoimmune diabetes[J].J Exp Med,1998,187(3):427-432.
[22] Tarhini A.Immune-mediated adverse events associated with ipilimumab ctla-4 blockade therapy:the underlying mechanisms and clinical management[J].Scientifica (Cairo),2013,2013:857519.
[23] Maker AV,Attia P,Rosenberg SA.Analysis of the cellular mechanism of antitumor responses and autoimmunity in patients treated with CTLA-4 blockade[J].J Immunol,2005,175(11):7746-7754.
[24] Jacobs JF,Nierkens S,Figdor CG,etal.Regulatory T cells in melanoma:the final hurdle towards effective immunotherapy?[J].Lancet Oncol,2012,13(1):e32-42.
[25] Chen L,Han X.Anti-PD-1/PD-L1 therapy of human cancer:past,present,and future[J].J Clin Invest,2015,125(9):3384-3391.
[26] Ortiz-Sanchez E,Helguera G,Daniels TR,etal.Antibody-cytokine fusion proteins:applications in cancer therapy[J].Expert Opin Biol Ther,2008,8(5):609-632.
[27] Kontermann RE.Antibody-cytokine fusion proteins[J].Arch Biochem Biophys,2012,526(2):194-205.
[28] Sathish JG,Sethu S,Bielsky MC,etal.Challenges and approaches for the development of safer immunomodulatory biologics[J].Nat Rev Drug Discov,2013,12(4):306-324.
[29] Teillaud JL.From whole monoclonal antibodies to single domain antibodies:think small[J].Methods Mol Biol,2012,911:3-13.
[30] Frankel SR,Baeuerle PA.Targeting T cells to tumor cells using bispecific antibodies[J].Curr Opin Chem Biol,2013,17(3):385-392.
[31] Wurch T,Pierre A,Depil S.Novel protein scaffolds as emerging therapeutic proteins:from discovery to clinical proof-of-concept[J].Trends Biotechnol,2012,30(11):575-582.
[32] Unciti-Broceta JD,Del Castillo T,Soriano M,etal.Novel therapy based on camelid nanobodies[J].Ther Deliv,2013,4(10):1321-1336.
[33] Coppieters K,Dreier T,Silence K,etal.Formatted anti-tumor necrosis factor alpha VHH proteins derived from camelids show superior potency and targeting to inflamed joints in a murine model of collagen-induced arthritis[J].Arthritis Rheum,2006,54(6):1856-1866.
[34] Silverman J,Liu Q,Bakker A,etal.Multivalent avimer proteins evolved by exon shuffling of a family of human receptor domains[J].Nat Biotechnol,2005,23(12):1556-1561.
[35] Beck A,Wurch T,Bailly C,etal.Strategies and challenges for the next generation of therapeutic antibodies[J].Nat Rev Immunol,2010,10(5):345-352.
[36] Stagg J,Galipeau J.Mechanisms of immune modulation by mesenchymal stromal cells and clinical translation[J].Curr Mol Med,2013,13(5):856-867.
[37] Bermudez-Humaran LG,Aubry C,Motta JP,etal.Engineering lactococci and lactobacilli for human health[J].Curr Opin Microbiol,2013,16(3):278-283.
[38] Farrar MD,Whitehead TR,Lan J,etal.Engineering of the gut commensal bacterium Bacteroides ovatus to produce and secrete biologically active murine interleukin-2 in response to xylan[J].J Appl Microbiol,2005,98(5):1191-1197.
[39] Rao S,Hu S,McHugh L,etal.Toward a live microbial microbicide for HIV:commensal bacteria secreting an HIV fusion inhibitor peptide[J].Proc Natl Acad Sci U S A,2005,102(34):11993-11998.
[40] Stephan MT,Moon JJ,Um SH,etal.Therapeutic cell engineering with surface-conjugated synthetic nanoparticles[J].Nat Med,2010,16(9):1035-1041.
[41] Kay MA.State-of-the-art gene-based therapies:the road ahead[J].Nat Rev Genet,2011,12(5):316-328.
[42] Perez EE,Wang J,Miller JC,etal.Establishment of HIV-1 resistance in CD4+T cells by genome editing using zinc-finger nucleases[J].Nat Biotechnol,2008,26(7):808-816.
[43] Holt N,Wang J,Kim K,etal.Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo[J].Nat Biotechnol,2010,28(8):839-847.
[44] Tebas P,Stein D,Tang WW,etal.Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV[J].N Engl J Med,2014,370(10):901-910.
[45] Burke BP,Boyd MP,Impey H,etal.CCR5 as a natural and modulated target for inhibition of HIV[J].Viruses,2014,6(1):54-68.
[46] 曹 玲,张 毅.嵌合抗原受体修饰T细胞治疗恶性肿瘤的研究进展[J].兰州大学学报(医学版),2015,41(1):1-8.
[47] Kalos M,June CH.Adoptive T cell transfer for cancer immun-otherapy in the era of synthetic biology[J].Immunity,2013,39(1):49-60.
[48] Brentjens R,Yeh R,Bernal Y,etal.Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells:case report of an unforeseen adverse event in a phase I clinical trial[J].Mol Ther,2010,18(4):666-668.
[49] Morgan RA,Yang JC,Kitano M,etal.Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2[J].Mol Ther,2010,18(4):843-851.
[50] Stronen E,Toebes M,Kelderman S,etal.Targeting of cancer neoantigens with donor-derived T cell receptor repertoires[J].Science,2016,352(6291):1337-1341.
[51] Rosenberg SA,Restifo NP.Adoptive cell transfer as personalized immunotherapy for human cancer[J].Science,2015,348(6230):62-68.
[52] Porter DL,Levine BL,Kalos M,etal.Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia[J].N Engl J Med,2011,365(8):725-733.
[53] Kalos M,Levine BL,Porter DL,etal.T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia[J].Sci Transl Med,2011,3(95):95ra73.
[54] Grupp SA,Kalos M,Barrett D,etal.Chimeric antigen receptor-modified T cells for acute lymphoid leukemia[J].N Engl J Med,2013,368(16):1509-1518.
[55] Maude SL,Frey N,Shaw PA,etal.Chimeric antigen receptor T cells for sustained remissions in leukemia[J].N Engl J Med,2014,371(16):1507-1517.
[56] Porter DL,Hwang WT,Frey NV,etal.Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia[J].Sci Transl Med,2015,7(303):303ra139.
[57] Rapoport AP,Stadtmauer EA,Binder-Scholl GK,etal.NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma[J].Nat Med,2015,21(8):914-921.
[58] Zhang T,Cao L,Xie J,etal.Efficiency of CD19 chimeric antigen receptor-modified T cells for treatment of B cell malignancies in phase I clinical trials:a meta-analysis[J].Oncotarget,2015,6(32):33961-33971.
[59] Zhang Y,Zhang T,Cao L,etal.Review of cancer immunotherapy:application of chimeric antigen receptor T cells and programmed death 1/programmed death-ligand 1 antibodies[J].Cancer Translational Med,2015,1(2):43.
[60] Gargett T,Yu W,Dotti G,etal.GD2-specific CAR T cells undergo potent activation and deletion following antigen encounter but can be protected from activation-induced cell death by PD-1 blockade[J].Mol Ther,2016,24(6):1135-1149.
[61] Liu X,Ranganathan R,Jiang S,etal.A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors[J].Cancer Res,2016,76(6):1578-1590.
[62] Kloss CC,Condomines M,Cartellieri M,etal.Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells[J].Nat Biotechnol,2013,31(1):71-75.
[63] Fedorov VD,Themeli M,Sadelain M.PD-1-and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses[J].Sci Transl Med,2013,5(215):215ra172.
[64] Pegram HJ,Lee JC,Hayman EG,etal.Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning[J].Blood,2012,119(18):4133-4141.
[65] Wu CY,Roybal KT,Puchner EM,etal.Remote control of therapeutic T cells through a small molecule-gated chimeric receptor[J].Science,2015,350(6258):aab4077.
[66] Cheng M,Chen Y,Xiao W,etal.NK cell-based immunotherapy for malignant diseases[J].Cell Mol Immunol,2013,10(3):230-252.
[67] Ames E,Murphy WJ.Advantages and clinical applications of natural killer cells in cancer immunotherapy[J].Cancer Immunol Immunother,2014,63(1):21-28.
[68] Lundqvist A,McCoy JP,Samsel L,etal.Reduction of GVHD and enhanced antitumor effects after adoptive infusion of alloreactive Ly49-mismatched NK cells from MHC-matched donors[J].Blood,2007,109(8):3603-3606.
[收稿2016-06-07]
(编辑 许四平)
10.3969/j.issn.1000-484X.2017.02.028
①本文受国家自然科学基金项目( 81171986,81271815,81502689)、河南省医学科技攻关计划项目(省部共建项目2015010004)、国家重点研发计划“重大慢性非传染疾病防治研究”重点专项(2016TFC1303500)和河南省重大专项(22130001)。
曹 玲(1985年-),女,在读博士,主要从事恶性肿瘤的免疫治疗研究,E-mail:caoling@gs.zzu.edu.cn。
及指导教师:黄 波(1969年-),男,博士,教授,博士生导师,主要从事肿瘤免疫、肿瘤生物学、肿瘤代谢等方面的研究,E-mail: tjhuangbo@hotmail.com。 张 毅(1964年-),男,博士,教授,博士生导师,主要从事肿瘤免疫治疗的基础研究与临床应用相关研究,E-mail: yizhang@zzu.edu.cn。
R392-33
A
1000-484X(2017)02-0288-09
②中国医学科学院基础医学研究所免疫学暨分子生物医学国家重点实验室,北京100005。
猜你喜欢
中国现代医生(2022年19期)2022-11-04
昆明医科大学学报(2021年2期)2021-03-29
科学导报(2021年4期)2021-02-22
文萃报·周五版(2021年2期)2021-01-25
临床肝胆病杂志(2020年2期)2020-12-14
中国现代医药杂志(2020年10期)2020-12-14
中国生殖健康(2020年7期)2020-12-10
天津医科大学学报(2019年3期)2019-08-13
医药前沿(2019年7期)2019-01-05
中国管理信息化(2016年23期)2017-02-04
