
Mn掺杂浓度对γ-TiAl合金延性影响理论研究
2017-04-14 05:22宋庆功蒋清杰顾威风胡雪兰
中国民航大学学报 2017年1期
宋庆功,蒋清杰,顾威风,胡雪兰
(中国民航大学a.理学院;b.中欧航空工程师学院,天津300300)
Mn掺杂浓度对γ-TiAl合金延性影响理论研究
宋庆功a,蒋清杰b,顾威风a,胡雪兰b
(中国民航大学a.理学院;b.中欧航空工程师学院,天津300300)
采用基于密度泛函理论的第一性原理方法,利用Materials Studio 6.0中的CASTEP模块对不同浓度的过渡金属元素Mn替位掺杂γ-TiAl得到的8个合金体系的原子平均形成能、晶格参量及轴比、弹性模量和重叠布居数等进行了计算分析。原子平均形成能的计算结果表明,各掺杂体系的形成能均为负值,表明其均可稳定存在,同时Mn原子占据Al原子位置得到的掺杂体系原子平均形成能更低,说明Mn原子倾向于替代Al原子;对晶胞轴比和弹性模量的计算分析表明,Mn掺杂能够提高合金的延性,以Ti12Al11Mn体系效果最为明显;重叠布居数的计算结果表明,Mn原子的掺入减弱了体系内共价键的各向异性程度,从化学键的角度解释了掺杂体系延性得到改善的机理。
掺杂γ-TiAl合金;延性;形成能;轴比;弹性模量;重叠布居数
TiAl基合金是新兴的一种金属间化合物高温结构材料,因具有高强度、低密度、高温抗蠕变以及抗氧化性质而备受青睐。随着航空业的发展,γ-TiAl基合金的应用受到高度重视[1-3],但γ-TiAl合金室温塑性差的缺点成为其在材料加工及应用中的瓶颈之一。因此,国内外的许多研究人员致力于γ-TiAl基合金的研究,以提升材料的室温塑性,并且发现合金化和微合金化是有效方法之一;采用非实验方法对材料结构和性能进行预测能够有效降低实验成本、缩短研发周期,效益显著。例如,王海燕等[4]采用第一性原理的方法对不同浓度的Mo元素掺杂γ-TiAl合金的力学性能进行了研究,发现Mo掺杂有利于提升γ-TiAl合金的塑性。宋庆功等[5-6]开展了4d过渡金属元素掺杂γ-TiAl合金的第一性原理研究,发现该类合金元素掺杂γ-TiAl合金的延性得到改善。
对于Mn元素掺杂γ-TiAl基合金的研究已有一些报道,掺杂可以有效抑制γ-TiAl合金的脆性。曹名洲等[7]通过实验制备出Mn掺杂γ-TiAl基合金,测试结果表明,合金的延性得到改善。李文等[8]采用理论方法计算分析了Mn掺杂γ-TiAl合金的价电子结构,从理论层面解释了其延性改善的部分机理。但是对于Mn掺杂的占位问题仍存在争议[9],掺杂浓度对其性质的影响也有待研究。因此,本文设计了4种浓度的Mn掺杂γ-TiAl基合金体系,采用基于密度泛函理论的第一性原理方法对其结构与性能进行了研究,以期探寻稳定的合金体系并预测其延性,为提升γ-TiAl基合金的延性提供理论依据。
1 体系结构模型和计算方案
1.1 结构模型
γ-TiAl合金的晶体空间构型为L10面心四方结构,空间群为P4/MMM,属于四方晶系,如图1(a)所示,其晶胞中含有2个Ti原子和2个Al原子,晶轴长度为a0=b0=3.98 Å,c0=4.04 Å,轴间交角为α=β= γ=90°[10]。γ-TiAl的最小结构单元为体心四方结构,如图1(b)所示,其晶格参量为a=b=2.832 Å,c=4.07 Å。

图1 γ-TiAl晶胞结构模型Fig.1 Structure models of γ-TiAl
为调节不同的掺杂浓度,本文以体心四方结构为基本单元,构建了2×2×2、3×2×2、3×3×2和3×3×3的γ-TiAl超胞体系,也就是Ti8Al8、Ti12Al12、Ti18Al18和Ti27Al27这4个体系。以这4个γ-TiAl合金超胞为基础,将一个Mn原子掺入其中,替位一个Ti原子或者Al原子,得到4种掺杂浓度的8个合金体系,即Ti7MnAl8、Ti8Al7Mn;Ti11MnAl12、Ti12Al11Mn;Ti17MnAl18、Ti18Al17Mn;Ti26MnAl27、Ti27Al26Mn。图2给出Mn原子替代Ti原子的4个掺杂体系,其对应的掺杂浓度分别为1/16、1/24、1/36和1/54(6.25at%、4.17at%、2.78at%和1.85at%);Mn替代Al原子掺杂体系与之类似。通过调节超胞的大小来控制Mn的浓度,该方法被广泛应用。研究表明,超胞的大小不同,会对计算结果产生微弱的影响,但不影响对于体系性质的分析[11-12]。
1.2 计算方案
本文采用Material Studio 6.0中的CASTEP模块对构建的纯γ-TiAl和Mn掺杂γ-TiAl体系进行计算处理。计算方案采用广义梯度近似下的PBE交换-关联函数;电子与离子间的交互作用采用超软赝势来描述。计算精度设置为Fine,以保证各体系的计算精度相同。CASTEP自选精度为:平面波截断能设置为310 eV;k点设置分别为4×4×3(16原子体系)、3×4×3(24原子体系)、3×3×3(36原子体系)以及3×3×2(54原子体系)。迭代计算收敛标准设置分别是:原子能量变化量小于1.0×10-5eV/原子;原子作用力为0.3 eV/nm;原子的位移小于0.001 Å;应力为0.05 GPa。根据这些设置,计算出每个纯体系和掺杂体系的晶格参量、形成能、轴比和重叠布居数等,并根据这些计算结果讨论分析各体系的稳定性和延性性质。根据以上参数设置完成后,将计算任务提交到96核计算机集群上进行计算,几何优化、能量性质和弹性模量的计算所需时间如表1所示。

图2 Mn替代Ti原子掺杂γ-TiAl基合金体系的结构模型Fig.2 Structure models of Mn-doped γ-TiAl systems
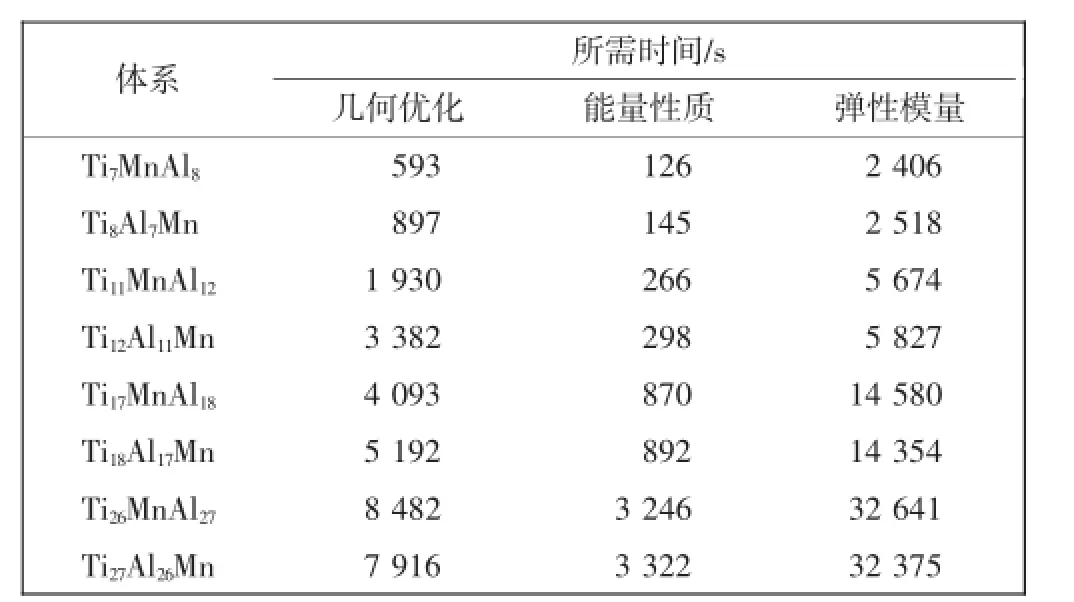
表1 计算所需时间Tab.1 Calculation duration
2 结果与分析
2.1 原子平均形成能
原子平均形成能是评价一个体系稳定性的重要指标。对于Mn掺杂γ-TiAl体系来说,原子平均形成能Ef可表示为

其中:Et是掺杂体系的总自由能;ETi、EAl和EMn分别是Ti、Al和Mn在单质结构中处于完全松弛状态下的单个原子能量;m和n分别是超胞中Ti原子和Al原子的个数;N表示掺杂对应超胞中原子总数。Ti、Al和Mn的单原子能量分别为-1 603.052 5 eV,-56.391 1 eV和-653.574 4 eV。
超胞中原子平均形成能越小,说明掺杂γ-TiAl体系的稳定性越高。各个浓度的Mn掺杂γ-TiAl体系和纯γ-TiAl体系的超胞自由能和原子平均形成能如表2所示。掺杂γ-TiAl体系的原子平均形成能均为负值,表明掺杂体系均可以制备并稳定地存在。同时,各掺杂超胞的原子平均形成能均大于对应的纯γ-TiAl体系的原子平均形成能,表明Mn掺杂会在一定程度上降低γ-TiAl的稳定性,这使得Mn掺杂γ-TiAl合金在实验制备上需要一定的实验条件,但是由于原子平均形成能的差距不大,可以尝试采用常规制备γ-TiAl合金的方法进行Mn掺杂制备,如真空自耗电弧熔炼(VAR)、真空感应熔炼之类的方法。随着Mn掺杂浓度的降低,掺杂体系的原子平均形成能逐渐变小,越来越接近纯体系的平均形成能。Mn掺杂浓度为1/54(1.85at%)的体系与纯γ-TiAl体系的原子平均形成能相差仅0.009 0 eV,表明该浓度的掺杂体系与纯γ-TiAl体系的稳定性相当,推测Mn低浓度掺杂γ-TiAl体系在实验中比较容易制备。
通过比较Mn占据Ti原子位置和Mn占据Al原子位置体系的原子平均形成能,可以发现Mn替代Al的体系原子形成能更低。这说明其更稳定,由此推测该类体系更容易制备。随着掺杂浓度的降低,Mn占据Ti位和占据Al位的原子平均形成能相差逐渐减小,说明Mn占据Al位和Ti位的几率越来越接近。事实上,Mn在掺杂γ-TiAl体系中的占位问题,一直存在争议[9]。从本文对于原子平均形成能的计算结果来看,Mn原子更倾向于占据Al原子的位置进行替位。
2.2 晶格参量和轴比
对纯和Mn掺杂γ-TiAl体系进行几何优化后的晶格参量如表3所示。为了便于直观地比较各体系的几何结构差异,已经将所有体系的晶格参量折算为γ-TiAl体系最小结构单元对应的量值。几何优化得到的纯γ-TiAl体系的晶格参量与文献报道的实验值相比[10],误差不超过1.25%,表明所选取的计算方法能够得到准确的晶格参量结果。

表2 Mn掺杂体系和纯体系的总能量和原子平均形成能Tab.2 Total energy and average atom formation energy of Mn-doped and pure γ-TiAl systems
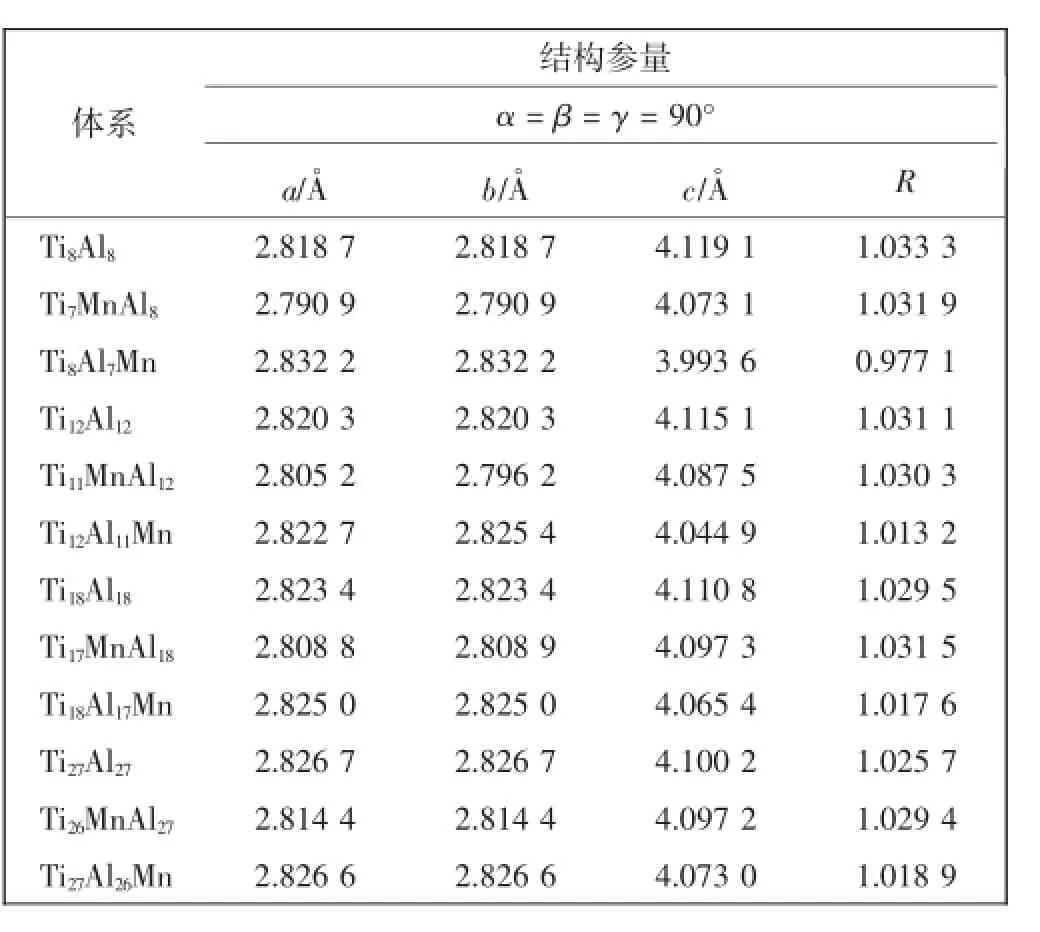
表3 Mn掺杂γ-TiAl体系与纯γ-TiAl体系的晶格参量和轴比Tab.3 Lattice parameters and axial ratio of Mn-doped and pure γ-TiAl systems
对各个Mn掺杂γ-TiAl体系的晶格参量进行对比分析,可发现对于Ti7MnAl8、Ti8Al7Mn、Ti17MnAl18、Ti18Al17Mn、Ti26MnAl27和Ti27Al26Mn 6个体系来说,其晶胞的a与b值均保持一致。而对于1/24原子掺杂浓度的Ti11MnAl12和Ti12Al11Mn 2个体系,晶胞a值和b值不再相等,分别产生了-0.009 0 A。和0.002 7 A。的差异,原因在于Mn原子的掺入,使2个体系的晶胞对称性发生了微小的改变。
对于掺杂γ-TiAl合金体系,晶胞的立方度是决定其延性的一个重要因素。对于L10面心四方结构,其立方度可用轴比(R=c0/a0)来描述。根据Kawabata等[13]的研究成果,R越接近于1,γ-TiAl合金的延性越好。为了便于比较,所有体系的a和c值已被转换成L10型面心四方晶胞的a0与c0量值,并且计算得到每个体系的超胞轴比,如表3所示。为了更加直观地分析Mn掺杂浓度对γ-TiAl体系轴比的影响和变化规律,通过图3给出了Mn掺杂γ-TiAl体系的轴比与掺杂浓度的关系。其中R1、R2各自代表Mn原子占据Ti原子位置掺杂体系和Mn占据Al原子位置体系的轴比。结果显示,相对于纯γ-TiAl体系,Mn原子占据Al原子位置对于轴比的改变量明显高于Mn占据Ti位的体系。随着掺杂浓度的上升,Mn替代Al原子体系的轴比逐渐下降,掺杂原子浓度为1/24的体系轴比最接近于1,预测其延性性质最好。还可以发现,Mn原子代替Al原子掺杂浓度在1/24~1/16(4.17at%~6.25at%)之间时,轴比最接近于1,所以推测Mn掺杂浓度在该区间时,γ-TiAl合金的脆性减弱、延性提升最明显。

图3 Mn掺杂浓度对γ-TiAl体系轴比的影响Fig.3 Effect of Mn concentration on axial ratio of doped γ-TiAl
2.3 弹性模量和延性
弹性模量是工程材料的一项重要的性能参数,其与材料的微观结构密切相关,是体系内原子或离子成键强度的宏观体现。弹性模量包括杨氏模量(Y)、剪切模量(G)和体积模量(B)等。根据Pugh等[14]的研究成果,由体积模量B和剪切模量G的比值B/G,可以判断出材料的延性:B/G的值小于1.75,表明材料为脆性材料;B/G的值大于1.75,表明材料具有较好的延性。纯γ-TiAl体系和各浓度Mn掺杂γ-TiAl体系的B与G值的计算结果如表4所示。纯γ-TiAl体系的B/G为 1.480,远小于1.75,表明其为脆性材料。分析各Mn掺杂γ-TiAl体系弹性模量可看出,Mn占据Al位掺杂体系的B值均大于Mn占据Ti位的B值,对于G值则相反。这使得Mn原子占据Al原子位置体系的B/G值大于占据Ti位的B/G值,也就是说,Mn原子替代Al原子体系的延性得到明显提升。从掺杂浓度来看,Mn替代Al原子浓度为4.17at%体系的B/G为1.751,使得体系的延性改善最明显,2.78at%和1.85at%掺杂浓度的B/G值次之。这些结论与前文对轴比的分析结果相一致。

表4 Mn掺杂γ-TiAl体系的弹性模量Tab.4 Elastic modulus of Mn-doped γ-TiAl
2.4 重叠布居数
文献[15]研究表明,TiAl合金的脆性与其内部共价键的强方向性有关,导致体系内部位错滑移时承受的派纳力较高,位错可动性差或者不可开动,造成TiAl合金脆性较大。本文计算了Ti12Al11Mn体系的化学键重叠布居数,并且与纯γ-TiAl的Ti12Al12体系进行了对比,结果如表5所示。对于纯γ-TiAl体系,Al原子之间存在较大的重叠布居数(0.39~0.67),表明Al原子之间有很强的共价结合作用,而Ti-Ti键和Ti-Al键的重叠布居数仅为0.14~0.16和0.12,共价结合作用明显弱于Al-Al键,这就导致各个化学键在不同的方向上具有很强的差异,导致γ-TiAl合金在宏观上表现出较强的脆性。而对于掺杂体系来说,相对于纯γ-TiAl体系,Al-Mn键布居数(0.36~0.45)小于纯体系中的Al-Al键,掺杂体系中的Al-Al键、Ti-Ti键和Ti-Al键布居数出现了多种量值,Al-Al键布居数总体呈下降态势,而Ti-Al和Ti-Ti共价键结合作用有所增强,表明各原子间共价键合的方向性有所降低,减弱了各向异性的程度,推测这是Mn掺杂γ-TiAl合金延性得到改善的原因之一。这与曲选辉等[16]通过实验观测压力作用下Mn掺杂γ-TiAl合金的位错滑移情况而得到的结论是一致的。

表5 Ti12Al11Mn和Ti12Al12体系的重叠布居数Tab.5 Overlap populations of Ti12Al11Mn and Ti12Al12
3 结语
本文利用基于密度泛函理论的第一性原理方法,研究了不同浓度的Mn元素掺杂对γ-TiAl合金体系性质的影响。分别建立了不同浓度的Mn元素掺杂γ-TiAl体系,通过计算Mn原子替代Ti和Al原子体系的原子平均形成能,发现Mn原子更倾向于替代Al原子。轴比计算表明,随着掺杂浓度上升,Mn替代Al原子掺杂体系的轴比逐渐下降,Mn掺杂浓度为4.17 at%的体系轴比最接近于1,推测其延性性质最好,而替代Ti原子体系的延性变化不明显。对于弹性模量和B/G的计算得到与对于轴比的分析结果相同的结论,两者互为佐证,表明4.17 at%浓度的Mn原子替代Al原子掺杂对于γ-TiAl合金的延性改善最有利。掺杂体系的重叠布居数计算结果表明,Mn掺杂降低了γ-TiAl合金体系中原子间共价键各向异性的程度,使得脆性有所降低。综上所述,Mn掺杂对于γ-TiAl合金的延性改善具有积极意义。
[1]CLEMENS H,MAYER S.Design,processing,microstructure,properties,and applications of advanced intermetallic TiAl alloys[J].Advanced Engineering Materials,2013,15(4):191-215.
[2]刘娣,张力军,米磊,等.TiAl合金的制备及应用现状[J].钛工业进展,2014,31(4):11-15.
[3]彭超群,黄伯云,贺跃辉.合金化对TiAl基合金性能的影响及机理[J].中国有色金属学报,1998,8(S1):11-15.
[4]王海燕,历长云,李旭升,等.Mo掺杂对TiAl合金物性影响的第一性原理研究[J].稀有金属材料与工程,2015,44(11):2737-2731.
[5]宋庆功,闫洪洋,康建海,等.4d过渡金属替位掺杂γ-TiAl基合金的第一性原理研究[J].材料导报,2014,28(1):150-154.
[6]宋庆功,秦国顺,杨宝宝,等.杂质浓度对Zr替位掺杂γ-TiAl合金的结构延性和电子性质的影响[J].物理学报,2016,65(4):046102-1-046102-9.
[7]曹名洲,韩东,周劲,等.含Mn的TiAl基合金的组织和性能[J].金属学报,1990,26(3):A223-A227.
[8]李文,蔡建岩,张瑞林.锰对TiAl脆性影响的价电子结构分析[J].有色金属,1998,50(4):96-98,89.
[9]李宝辉,孔凡涛,陈玉勇,等.TiAl金属间化合物的合金设计及研究现状[J].航空材料学报,2006,26(2):72-78.
[10]陈国良,林均品.有序金属间化合物结构材料物理金属学基础[M].北京:冶金工业出版社,1999:285-286.
[11]刘显坤,刘颖,郑州,等.Ti及氢化物几何与电子结构的密度泛函理论研究[J].稀有金属材料与工程,2010,39(5):832-837.
[12]侯清玉,赵春旺,李继军,等.Al高掺杂浓度对ZnO导电性能影响的第一性原理研究[J].物理学报,2011,60(4):047104-1-047104-6.
[13]KAWABATA T,TAMURA T,IZUMI O.Effect of Ti/Al ratio and Cr, Nb,and Hf additions on material factors and mechanical properties in TiAl[J].Metal Transaction,1993,24 A(1):141-150.
[14]PUGH S F.Relation between the elastic moduli and the plastic properties of polycrystalline pure metals[J].Philosophical Magazine,1954, 459(367):823-843.
[15]孔凡涛,陈子勇,田竞,等.提高TiAl基合金室温塑性的方法[J].稀有金属材料与工程,2003,32(2):81-86.
[16]曲选辉,黄伯云,吕海波,等.合金化对TiAl有序金属间化合物室温脆性变形的影响[J].稀有金属材料与工程,1993,22(1):11-16.
(责任编辑:杨媛媛)
Effects of impurity concentration on ductility of Mn-doped γ-TiAl alloys
SONG Qinggonga,JIANG Qingjieb,GU Weifenga,HU Xuelanb
(a.College of Science;b.Sino-European Institute of Aviation Engineering,CAUC,Tianjin 300300,China)
Using the first-principle method based on density functional theory,Mn-doped γ-TiAl alloy systems,of which the concentrations of Mn substituting Ti or Al are 1/54,1/36,1/24 and 1/16 respectively,have been investigated with CASTEP aimes at improving its ductility.Meanwhile,the structure,energy,plastic and electronic properties are calculated and analyzed.The average atom formation energy of all the doped systems is negative,indicating the energy stability of each system.The Mn-substituting-Al systems have got a lower formation energy,which shows that the Mn tends to substitute Al.Results of both the axial ratio and elastic modulus show that doping with Mn can improve the ductility of γ-TiAl alloys by substituting Al,with Ti12Al11Mn showing the best ductility.Comparison of the overlap population of Ti12Al11Mn and pure γ-TiAl indicates doping with Mn can weaken the anisotropy of the covalent bonds in γ-TiAl systems,which contributes partly to the ductility improvement of γ-TiAl alloys.
doped γ-TiAl alloy;ductility;formation energy;axial ratio;elastic modulus;overlap population
TG146.2
A
1674-5590(2017)01-0060-05
2016-04-10;
2016-05-12基金项目:国家自然科学基金项目(51201181)
宋庆功(1958—),男,河北唐山人,教授,博士,研究方向为新型材料设计与制备.
猜你喜欢
湖南交通科技(2021年1期)2021-04-28
安徽建筑(2020年3期)2020-04-17
地震研究(2019年4期)2019-12-19
中国航海(2019年3期)2019-10-30
表面技术(2019年6期)2019-06-27
有色金属材料与工程(2016年6期)2017-05-31
北京航空航天大学学报(2017年12期)2017-04-23
汽车文摘(2015年8期)2015-12-15
现代防御技术(2015年3期)2015-05-05
有色金属材料与工程(2013年1期)2013-12-26
