
Modification of CaO-based sorbents prepared from calcium acetate for CO2 capture at high temperature☆
2017-05-28 10:22XiaotongLiuJunfeiShiLiuHeXiaoxunMaShisenXu
Xiaotong Liu ,Junfei Shi,Liu He ,Xiaoxun Ma ,*,Shisen Xu
1 School of Chemical Engineering,Northwest University,Xi'an 710069,China
2 Chemical Engineering Research Center of the Ministry of Education for Advanced Use Technology of Shanbei Energy,Xi'an 710069,China
3 Shaanxi Research Center of Engineering Technology for Clean Coal Conversion,Xi'an 710069,China
4 China Huaneng Group Clean Energy Technology Research Institute,Beijing 100098,China
1.Introduction
CO2is the main contributor to the greenhouse effect,which has attracted increasing attention all over the world.Therefore,to effectively reduce CO2emission is urgently needed.The key for this problem is to control and capture the CO2from fossil fuel power plants,which contributes one-third of the anthropogenic CO2emissions[1,2].
The current CO2separation technologies include absorption,adsorption,membrane separation,and cryogenic separation[3–6].However,these technologies could hardly be conducted until the high temperature flue gas was cooled below 100°C or even at room temperature.A huge of energy was consumed in the cooling period,equal to about 13%–37%of net generation[7].Therefore,it is important to develop the high temperature sorbent to capture CO2.
With the advantages of wide sources,low cost,high adsorption capacity,easy preparation and regeneration,CaO-based sorbents become the promising candidate for capturing CO2at high temperature[8–10].Recently,Heiko and colleagues[11–13]reported that calcium looping has been successfully implemented the process demonstration at pilot scale,which have been erected at IFKin Germany(200 kW),Technische University Darmstadt in Germany(1 MW),la Pereda in Asturias—Spain(1.7 MW)etc.In the carbonator of dual fluidized bed system(DFB)(carbonator,650 °C;regenerator,900 °C),the CO2adsorption efficiency of limestone could be kept above 85%for long periods,such as 22 h.
In spite of low cost,the adsorption capacity of limestone dropped dramatically with the increasing of cycle number due to the sintering at high temperature[14,15],which is the general disadvantage for CaO-based sorbents.A mass of fresh sorbents should be added into carbonator to maintain the adsorption stability due to the poor cyclic stability of CaO-based sorbents like limestone.Therefore,the more frequent regeneration resulted in high cost.For this problem,a lot of researchers attempted to improve the cyclic stability of CaO-based sorbents by doping elements.The sorbents doped with different elements included CaO/Ca12Al14O33(75:25,mass ratio)[16],nano-CaCO3/SiO2(39:1,mole ratio)[17],CaCO3/Mn(NO3)2(100:1,mole ratio)[18],CaTi-50[19],Longcliffe limestone/HBr(0.167,mole ratio)[14],and so on.The carbonation conversions(mol-CO2/mol-CaO)of corresponding sorbents after several carbonation/calcinations respectively were 70%(50th cycle),27%(20th cycle),32%(18th cycle),45%(27th cycle)and 25%(13th cycle),while the carbonation conversions of corresponding fresh sorbents were 85%,37%,73%,99%and 100%,respectively.
The cyclic stability of sorbents could be improved by doping elements.However,the decay was still obvious even the cycle number was only about 20,which was the general number to test the cyclic stability in most previous researches.
As a participant of the international cooperation project of China and the United States,our group undertook the subtask,research and development of solid sorbents for high temperature CO2adsorption.The CO2sorbent was needed to prepare to be a candidate for demonstrative project of Huaneng IGCC power plant in Tianjin.In the previous work,different precursors for CaO had been studied,such as CaCO3,CaC2O4,Ca(OH)2and CaAc2[20].The adsorption capacity of CaO derived by CaAc2is significantly large due to the porous structure generated by calcination.It was also found that there was a certain degree of increase for cyclic stability with doping element,especially for that with the doping ratio of 8:2,among 9:1,8:2,7:3 and 6:4[21–24].
The durability of sorbent is an important indicator of industrial application.The more stable adsorption capacity of sorbents resulted in the less added fresh sorbents.Then,the operating cost was reduced.However,such a short cycle number(such as 20)was not long enough to show the decay trend of adsorption capacity.
The objective of the present work is to modify the CaO derived from CaAc2(CaO–CaAc2)by doping different elements(Mg,Al,Ce,Zr and La)to strengthen the channel structure of CaO and restrain sintering.The cyclic stability was studied by increasing the cycle number so as to explore more stable sorbents for the possibility of industrial application.Meanwhile,the effects of specific surface area and channel structure on adsorption capacity with the increase of carbonation/calcination cycles were investigated.
2.Experimental
2.1.Sorbent preparation
All the reagents used in present study were analytically pure.The CaO was derived from CaAc2.The doped elements were respectively Mg,Al,Ce,Zr and La,which derived from the corresponding nitrates.The precipitant was ammonia water(Tianjin Tianli Chemical Co.,China,500 ml,25%–28%).The target doping mole ratio of Ca and the doping element was 8:2.Taking an example of Mg,the preparation was as follows.The quantitative CaAc2·H2O(Chengdu Fuchen Chemical Co.,China,250 g,>98.0%)and Mg(NO3)2·6H2O(Chengdu Kelong Chemical Co.,China,500 g,99.0%)were dispersed in the distilled water and stirred at 80°C.The excess ammonia water was added to generate magnesium hydroxide.Then,the distilled water was evaporated.The absolute ethyl alcohol(Tianjin Fuyu Chemical Co.,China,500 ml,>99.7%)was added as the solvent.The slurry was stirred overnight and dried at120°C to obtain a solid sample.Then,the sample was calcinated in the muffle furnace at900°C for 5h before the carbonation test in TGA,which was called pre-calcination in this paper.The sample was composed of calcium oxide and magnesium oxide,which was marked as CaO–Mg.
2.2.TGA experiments
The carbonation experiments were conducted with a STA449F3 thermogravimetric analyzer(TGA).The samples,about 10 mg,were calcinated for 30 min at 700 °C under nitrogen(70 ml·min−1,99.99%)with a heating rate of 20 °C·min−1.Then,the adsorption capacities of sorbents were gotten when CO2(99.99%)with 30 ml·min−1was introduced to the system as long as 40 min.As the slight quantities of samples,there was slight exothermic in TGA compared with DFB in practical pilot scale.A higher carbonation temperature was selected to increase the carbonation conversion.The lower calcination temperature helped to retard inactivation of sorbents and reduce energy consumption,which still made the sorbent regenerate completely.
For the carbonation/calcination cycle experiments,the carbonation was conducted at 700 °C for 9 min with N2(70 ml·min−1)and CO2(30 ml·min−1).The calcination was followed at the same temperature and kept for 20 min without CO2.In cycling tests,the carbonation time was selected as 9 min because the sorbents had already been almost totally carbonated.
In present work,the oxides of doping elements were considered as inert support,which could not react with CO2in the experiment conditions.The carbonation conversion(X)of sorbents was defined as the mole ratio of the adsorbed CO2and the effective CaO of sorbent.Taking an example of doping Mg,the carbonation conversion of CaO–Mg was de fined asXMg(8:2)shown as Eq.(1).

m0is the mass of calcined sorbent recorded by TGA before carbonation.mtis the real-time mass of sorbent at any given moment while carbonation.mt−m0means a mass gain due to CO2uptake by sorbent when the carbonation carries on thetminute.αMg(8:2)is a coefficient to calculate the effective mass of CaO in CaO–Mg as Eq.(2).
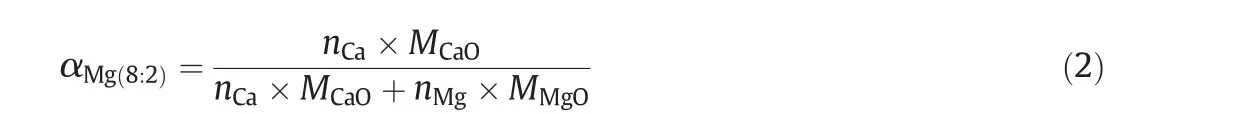
nis the mole number,andMis the molar mass.nCa/nMgis 8:2.
The multiple cycle stability is signed asSN,shown in Eq.(3),which is the ratio of the carbonation conversions for theNth cycle and the 1st cycle,andNis the number of the cycle.The cyclic stability of CaO–Mg through 22 cycles could be marked asS22(CaO–Mg).

2.3.Sorbent characterization
The pore structure parameters of the calcined samples were examined from N2adsorption and desorption isotherms at the temperature of liquid N2(QUANTACHROME AUTOSORB-1).The specific surface area and pore volume were calculated from the Brunauer Emmet Teller(BET)equations and Barrett–Joyner–Halenda(BJH)method,respectively.The microstructure of calcined samples was investigated by field emission scanning electron microscopy(SEM;Zeiss Supra 55(VP))with 3 kV of accelerating voltage under high vacuum.The phase composition of samples was determined by X-ray diffraction(XRD;Smartlab and EMPYREAN)with Cu Kαradiation,λ =0.1542 nm in the 2θ range of 10°–80°with a scanning step of 0.01°.
3.Results and Discussion
3.1.Effect of doped element on carbonation conversion
In Fig.1,the TGA data showed that the carbonation conversion of CaO–CaAc2was about 94.1%after 40 min carbonation.Obviously,CaO–CaAc2was almost totally carbonated after 40 min carbonation.In the process of calcinations for CaAc2(Eqs.(4)–(6)),the escape of a variety of gas by the multi-step decomposition formed the porous lamellar structure as shown in Fig.2(a).The uniform,disperse and loose CaO particles were conductive to the adsorption performance[25,26].As shown in Table 1,the specific surface area and pore volume of fresh CaO–CaAc2were relatively large.Therefore,the diffusion resistance of CO2decreases and the chance for CO2reacting with CaO inside the particles increases.

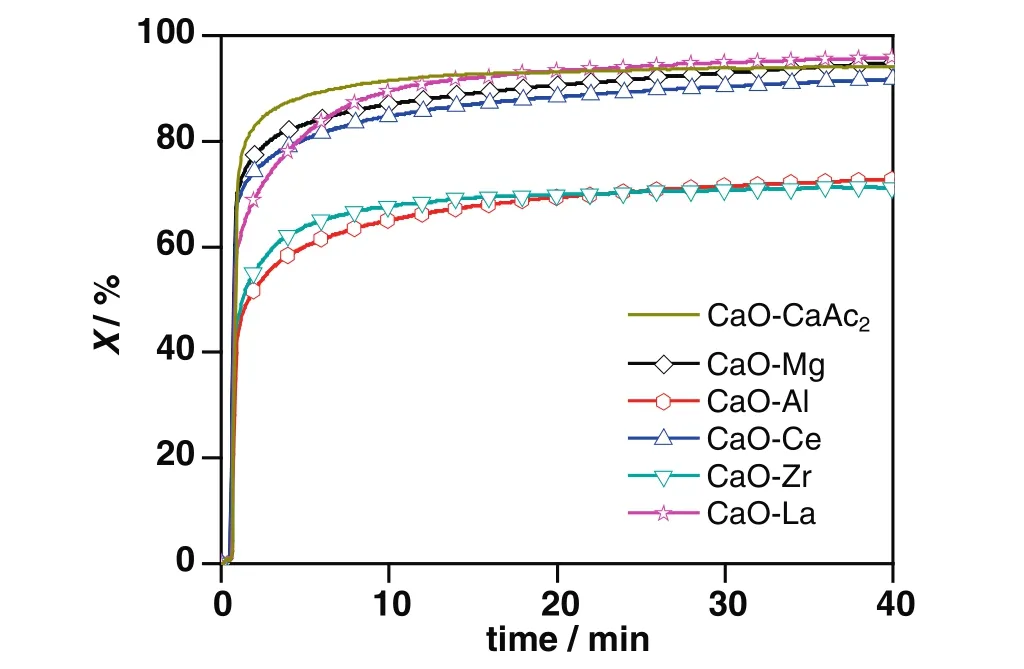
Fig.1.Carbonation conversions of sorbents doped with different elements during 40 min carbonation.Conditions:carbonation temperature,700°C;CO2 concentration,30 vol%;balance nitrogen.
With the doping ratio of 8:2,there was little improvement for carbonation conversions of modified sorbents during 40 min carbonation(Fig.1).The only exception was CaO–La,the carbonation conversion of which was as high as 95.8%after 40 min carbonation.La(OH)3produced in the process of preparation could be decomposed into La2O3as Eqs.(7)and(8)[27]at 900°C in muffle furnace.La2O3could react with CO2to generate La2O2CO3[28]as Eq.(9).As shown in Fig.3,there were respectively La2O3and La2O2CO3in CaO–La before and after 40 min carbonation by XRD.By assuming thatthe doped La existed in the form of inert La2O3in calculation,the active La2O3in CO2capture resulted in a larger calculated carbonation conversion for CaO–La than other inert doping sorbents.

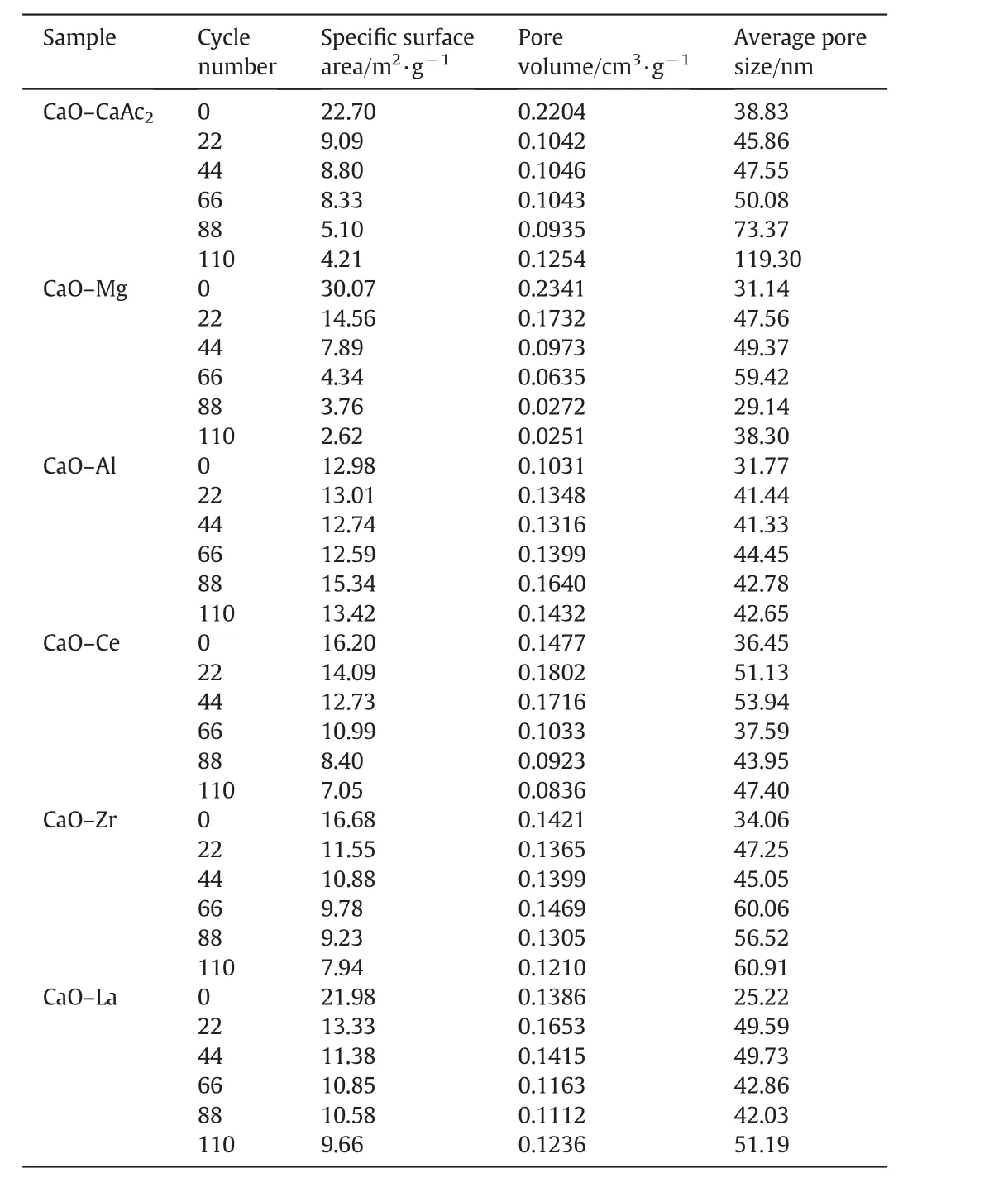
Table 1The BET data of sorbents after different number of cycles

For CaO–Mg and CaO–Ce,XRD shows that the doped elements always respectively existed in the inert form of MgO and CeO2before and after carbonation.Only CaO of CaO–Mg and CaO–Ce contributed the CO2uptake.Compared with CaO–Ce,the greater carbonation conversion of CaO–Mg was bene fit from the large specific surface area and pore volume(Table 1),which increased the contact chance for CO2.Therefore,the diffusion resistance for CO2reduced.

Fig.2.SEM of CaO–CaAc2 before and after 22 cycles:(a)fresh CaO–CaAc2;(b)CaO–CaAc2 after 22 cycles.
In the initial 9 min carbonation,the carbonation conversions of modified sorbents were much lower than that of CaO–CaAc2as shown in Fig.1,which was the result of the slight sintering by calcinations in the preparation stage.With the different initial carbonation conversion rates,the difference of carbonation conversions after 40 min carbonation for CaO–La,CaO–Mg,CaO–Ce and CaO–CaAc2was little.It meant that the diffusion in a relatively short carbonation time could affect the adsorption capacities of sorbents.When the carbonation time was long enough,the sorbents would be totally carbonated.
However,the carbonation conversions of CaO–Al and CaO–Zr after 40 min carbonation were much less.The poor carbonation conversions of CaO–Al and CaO–Zr were due to the loss of CaO rather than the relatively small specific surface area.For CaO–Al,there was Ca12Al14O33(Fig.3)generated as Eq.(10)[16,29]in muffle furnace during the precalcination period,which consumed a part of the effective CaO.Then,the calculated carbonation conversion of CaO–Alwas less than the actual one,which was also much less than that of CaO–CaAc2.As the presence of CaZrO3shown in XRD from Fig.3,the worst carbonation conversion of CaO–Zr could be considered as the result of the sacrifice of some effective CaO by reacted with ZrO2as Eq.(11)[30].


Fig.3.XRD of sorbents with different doping elements:(a)fresh sorbents,(b)sorbents after 40 min carbonation.

3.2.Effect of doped element on cyclic CO2
As the durability was paid more attention in industry,the sorbents were tested through 22 carbonation/calcination cycles at 700°C(Fig.4).Compared with the dramatically decreased carbonation conversion for CaO–CaAc2with the increasing of cycle number,the cyclic stabilities of modified sorbents were improved in varying degrees.
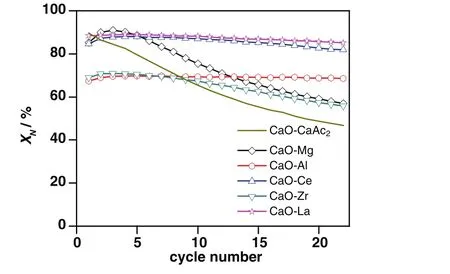
Fig.4.Carbonation conversions of sorbents through 22 carbonation/calcination cycles.
The carbonation conversions of CaO–Mg,CaO–Ce and CaO–La in the 1st cycle were close to that of CaO–CaAc2,about 95%.With the increase of cycle number,the decay of carbonation conversions for CaO–Ce and CaO–La was very slight.Through 22 cycles,the carbonation conversion of CaO–La was the greatest.XLa(8:2)was as high as 85.1%in the 22nd cycle,which might be due to the extra reaction of La2O3and CO2.The migration of lattice oxygen could help to maintain the high activity for sorbent to capture CO2.S22(CaO–Ce)was 96.7%.Through the 22 cycles,the carbonation conversion of CaO–Ce was always a little less than that of CaO–La.Although CaO–Mg showed the largest decay amplitude of carbonation conversion,S22of CaO–Mg was far better than that of CaO–CaAc2.As the sacrifice of some effective CaO,the carbonation conversions of CaO–Al and CaO–Zr in the 1stcycle were poor.However,the sharp decay of carbonation conversion for CaO–CaAc2slowed down with the doping Zr.After the 9th cycle,XZr(8:2)was largerthanXCaO–CaAc2.Both the carbonation conversion and cyclic stability of CaO–Zr were higher than those of CaO–CaAc2after 22 cycles.It was worth noting thatXAl(8:2)was 68.6%after 22 cycles,which was kept as high as the one in the 1st cycle.
As the Tammann temperature of CaCO3is 533°C[30],the CaCO3generated by the carbonation reaction might begin to melt.All the atoms in the lattice began to loose at 700°C for a long time.Therefore,the loose channels of CaO–CaAc2were easily changed.Some channels were amalgamated into irregular large orifices and others disappeared after multiple cycles(Fig.2(b)).As shown in Table 1,the pore volume decreased significantly and the average pore size became larger after 22 cycles.The specific surface area after 22 cycles was only left 40%of the original one.Then,CO2diffusion resistance increased.The carbonation conversion of CaO–CaAc2was sharply decreased with the increasing of the cycle number.With the worst cyclic stability through 22 cycles in the five kinds of modified sorbents,the decay of specific surface area of CaO–Mg was the largest.The proportion of mesoporous with larger pore size increased.However,the pore volume of CaO–Mg decreased much slower than that of CaO–CaAc2,which meant that the porosity of CaO–Mg was not changed significantly.Table 1 shows that the specific surface area and pore volume of all the modified sorbents after 22 cycles was maintained better than that of CaO–CaAc2.The doped element could help to get the more stable skeletal structure[31]and retard the sintering,which improved the cycle stability of modified sorbents.
Different from the monotonic decreasing of carbonation conversion for CaO–CaAc2,the carbonation conversions of modified sorbents increased slightly in initial 4 cycles.It was found that there was a selfreactivation phenomenon[31,32]when the lost specific surface area could be compensated due to the formation of a hard skeleton by doped element,which could isolated the contact of melting CaCO3in some degree.The sintering was retarded in varying degrees.
3.3.Effect of doped element on extended cyclic CO2
Since 22 cycles was not long enough to identify the difference of the sorbents,the cycle number was increased to 110 to research the cyclic stability.Since it is inappropriate for equipment maintenance of TGA to keep a continuous long time at high temperature,the 110 carbonation/calcination cycles were conducted by repeating 5 times the 22-cycle experiments.
Fig.5 shows the cyclic performance of sorbents through 110 cycles.The carbonation conversions of all sorbents decreased with the increasing of cycle number.Compared with CaO–CaAc2,the decay trends of carbonation conversions for the modified sorbents became slow.After 110 cycles,allX(8:2)were larger thanXCaO–CaAc2.The decay trends of carbonation conversions for CaO–Mg and CaO–Zr were similar with CaO–CaAc2.
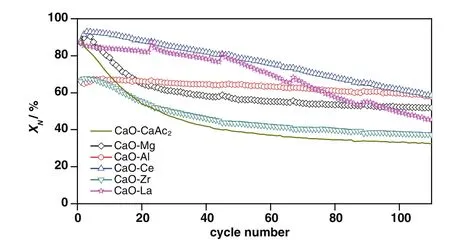
Fig.5.Carbonation conversions of sorbents through 110 carbonation/calcination cycles.
The carbonation consisted of chemical reaction control stage and diffusion control stage,which also called rapid reaction control stage and slow reaction control stage[33,34].As the reports,the diffusion control stage started when the product layer thickness of CaCO3was more than 49 nm[35,36].The product layer could obstruct the contact between CO2and the internal CaO.With larger molar volume(36.9 cm3·mol−1),the generated CaCO3might block the narrow channels.Then,it was difficult for CO2to diffuse into the core of CaO(16.9 cm3·mol−1[10,37])particles.The critical point occurred in the end of chemical reaction control stage and the beginning of diffusion control stage.In the report of Shiet al.[38],the critical point was de fined as two times of the minutes when the maximum adsorption rate appeared.In fact,the peaks in our experiments might be double and irregular.After the maximum adsorption rate appeared,there was a time when the increasing rate of carbonation conversion fell below 15.This time was identified as the critical time by our team(Fig.6),which was signed astcin every cycle.As the rapid reaction control stage was not started as soon as the beginning of every cycle,the retardation time,tr,was de fined as the time between the beginning of every cycle and the time when the increasing rate of carbonation conversion was above 15.The difference value oftcandtrwas the actual time of rapid reaction control stage in theory,which was marked astf.
The averagetcof modified sorbents was greater than that of CaO–CaAc2through 110 cycles(Table 2).For all sorbents,the carbonation conversion in chemical reaction control stage contributed to about 80%of total carbonation conversion in 9 min(Fig.7).Then,the increasing rate slowed down due to the change of pore structure for sorbents.Althoughtrincreased with the increasing cycle number in every group,the averagetrof every group was similar.Then,the decay trends of averagetfandtcwere the same.The average critical time of every group was proportional to the corresponding carbonation conversion.The results were consistent with the found that the carbonation conversion in chemical reaction control stage is linearly proportional to the BET specific surface area[39].The decay trend ofXtwith the increase of cycle number in Fig.7 was in accordance with the change of specific surface area in Table 1.The critical time shows the adsorption efficiency of sorbents,which indicates the appropriate carbonation time in experiments.The adsorption time could be reduced according to the critical time,which could save the experiment time and delay the sintering of sorbents.
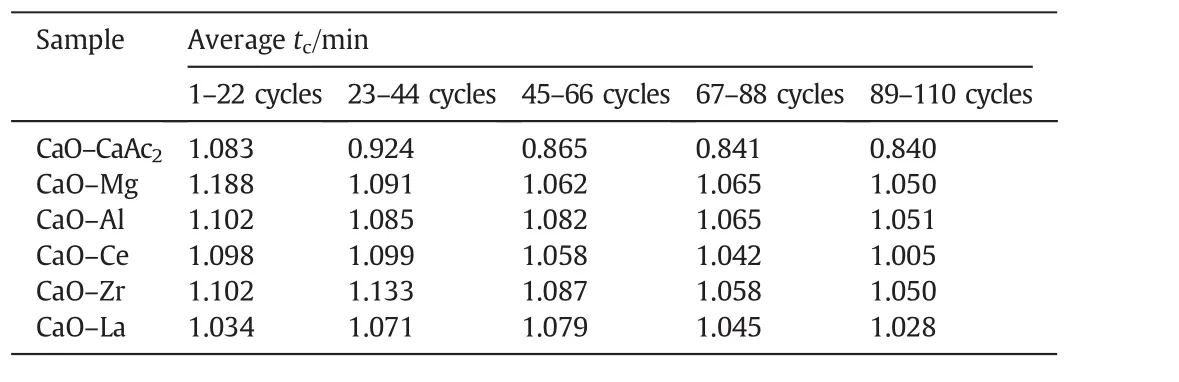
Table 2The average critical time of sorbents in 5 groups of 22-cycle through 110 cycles
For CaO–Mg and CaO–Zr,there was about 80%of decay of carbonation conversion occurred in initial 44 cycles.The main decay oftfandtcalso occurred in initial 44 cycles as shown in Fig.8.There was obvious decay of specific surface area and the pore volume in mesoporous with small pore size in initial 44 cycles shown in Table 1 and Fig.9.As the channels gradually narrowing due to the carbonation of CaO[40],it was more difficult for CO2to diffuse to the core of CaO through narrow channels than the wide ones.Therefore,the smaller pore size was beneficial to specific surface area but against the mass transfer and CO2diffusion.When the pore size was large,CO2could diffuse more easily.However,the integration of channels reduced the specific surface area of sorbent,which was not conducive to contacting and reacting with CaO for CO2.Compared with the decay trend of carbonation conversion and the change of pore size distribution,the more mesoporous with small pore size,about 2 nm to 8 nm,was benefit for the carbonation.
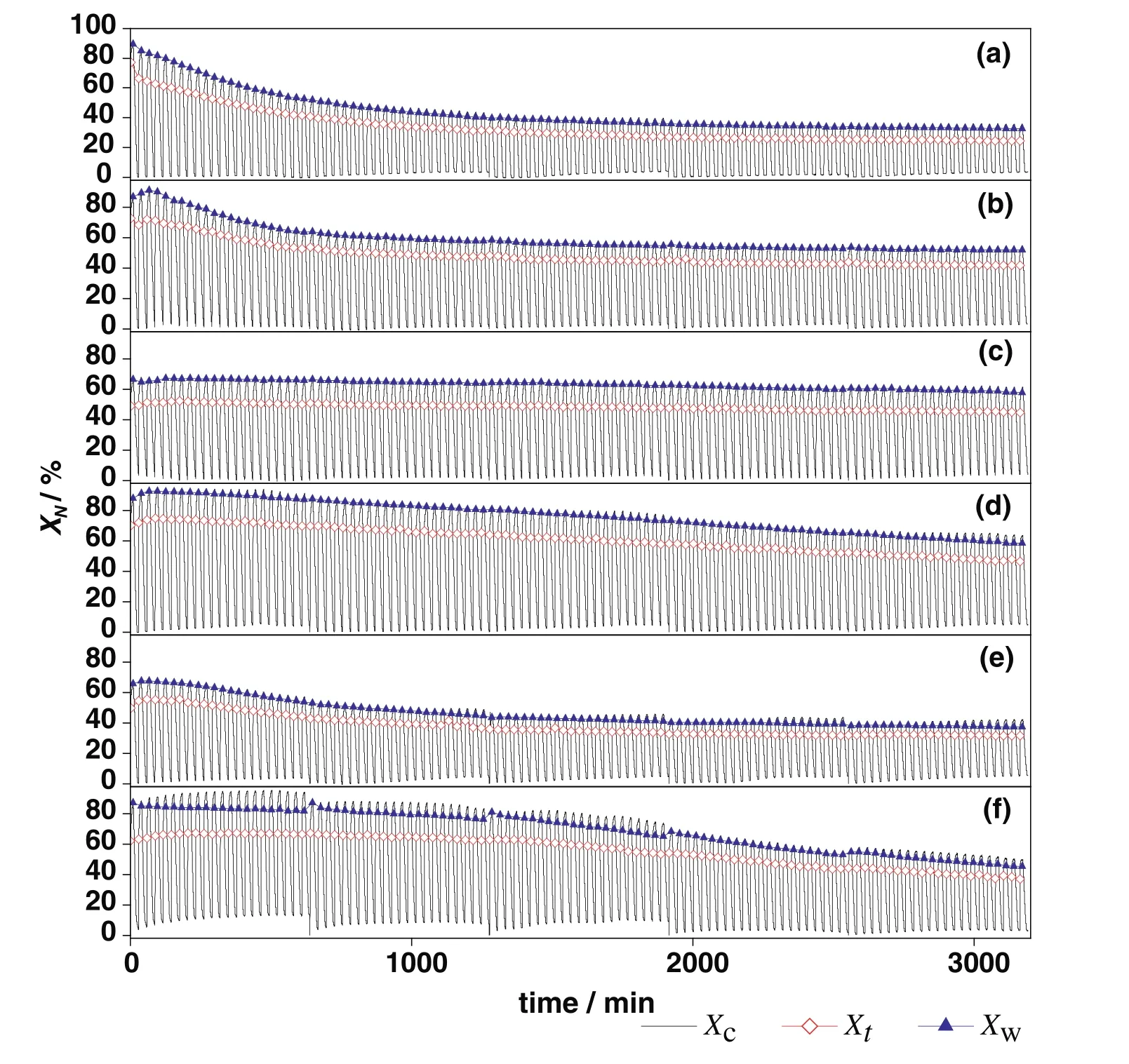
Fig.7.The 110-cycle comparison of carbonation conversions ofsorbents:(a)CaO–CaAc2,(b)CaO–Mg,(c)CaO–Al,(d)CaO–Ce,(e)CaO–Zr,(f)CaO–La.(X c was the carbonation conversion in whole continuous cycles.Among them,Xt was the carbonation conversion in t c of every cycle,and X w was the carbonation conversion in 9th-min-carbonation of every cycle.)
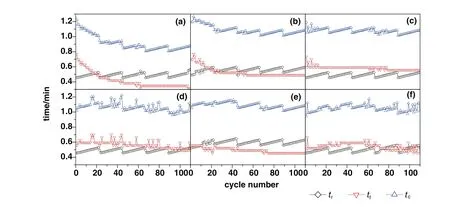
Fig.8.The retardation time(t r),actual time of rapid reaction control stage(t f)and critical time(t c)of sorbents with the number of cycles:(a)CaO–CaAc2,(b)CaO–Mg,(c)CaO–Al,(d)CaO–Ce,(e)CaO–Zr,(f)CaO–La.
Thetcof CaO–Mg and CaO–Zr(Table 2)became much shorter with the increase of cycle number,which meant that the time of chemical reaction control stage became much shorter and the conversion rate in this stage would decrease.At the same time,the diffusion control stage played a more and more important role in carbonation conversion.With the shorter time in chemical reaction control stage and the longer time in diffusion control stage,the increasing rate of carbonation conversion would be reduced.Therefore,the carbonation conversions of CaO–Mg and CaO–Zr decreased with the increasing of cycle number due to the decrease of carbonation conversion in both chemical reaction control stage and diffusion control stage.
界面态位于靠近Si-SiO2界面1~2个原子键约0.5 nm的距离处,可以较快地与硅导带和价带交换电荷,促进了电子通过热运动由价带跃迁到导带,导致表面有效产生速度se增大。由式(1)可知,se增大,Js将增大。电子辐射引起的位移效应,在像素单元体耗尽区内产生大量的体缺陷,这些缺陷能级在禁带中起到产生-复合中心的作用,使耗尽区载流子寿命τg显著减小,由式(2)可知,τg显著减小,Jg将显著增大。
Compared with CaO–CaAc2,the skeleton supporting structure made by MgO or CaZrO3was more effective to retard the rapid decay of CO2adsorption capacity.With the doping element,the pore size distribution in mesoporous became stable(Fig.9).After 110 cycles,even with irregular large orifices and cracks left on the surface of particles(Fig.10),the carbonation conversions of CaO–Mg and CaO–Zr were much greater than that of CaO–CaAc2.
Even with the remarkableS22,the carbonation conversions of CaO–Ce and CaO–La decreased obviously with the extended cycle number.For the last 4 group of 22-cycle,the decay of carbonation conversions for CaO–Ce and CaO–La were respectively at almost uniform rate in the every group.With the increase of cycle number,the sintering gradually occurred.XCe(8:2)dropped to 58.5%after 110 cycles.However,the sintering of CaO–Ce was greatly slowed down compared with CaO–CaAc2.The specific surface area of CaO–Ce through 110 cycles was decreased at a relatively slow and uniform rate.The SEM of CaO–Ce through 110 cycles showed that there were still abundant uniform pores on the surface of the particle.Therefore,the carbonation conversion of CaO–Ce through 110 cycles was always much greater than others.For CaO–La,the decay was more obvious in the last several cycles.The carbonation conversion of CaO–La after 110 cycles was only 45.4%.Even with several irregular data,Fig.8 shows thattfandtcdecreased slightly in initial cycle number.The most decay oftcoccurred in the last 44 cycles shown in Table 2.It was difficult to find the skeletal structure on the surface of CaO–La in Fig.10(f)due to the block of molten CaCO3.There were only cracks and tiny holes left.The specific surface area of CaO–La was still kept relatively large after 44 cycles(Table 1),which was assumed that the integration of channels mainly occurred in the subsequent cycles.Fig.9 verified that the most decrease of pore volume in mesoporous with smaller pore size occurred in the last 66 cycles.In the report[28],the cyclic stability of the La-doped sorbent could be kept as high as 75%after 50 cycles.Although the decay was relatively sharp in the extended cycles,SCaO–Lawas still about 75%through 70 cycles.As a result of the doped Ce or La,the amplitude of variation of pore size distribution through 110 cycles was much fewer than that of CaO–CaAc2.In terms of existing 110 cycles,CaO–Ce and CaO–La showed better adsorption capacity and cyclic stability.
As shown in Fig.10(c),the surface microstructure of CaO–Al after 110 cycles was the most perfectly maintained.There was no obvious decay of carbonation conversion with the increase of cycle number.X(8:2)of CaO–Al was 57.9%after 110 cycles while the carbonation conversion of fresh CaO–Al was 66.5%.S110(CaO–Al)was as high as 87.1%.tfandtcfor CaO–Al was more stable than other sorbents.There was no obvious decrease of carbonation conversion in either chemical reaction control stage or diffusion control stage(Fig.7(b)).The specific surface area(Table 1)and pore size distribution(Fig.9(c))kept constant through different cycles,which meant that the channel structure of sorbent was stable.Consistent with the adsorption performance of sorbents,CaO–Al kept the most stable skeletal structure and abundant uniform fine channels in the SEM of modified sorbents after 110 cycles.Because Ca12Al14O33could form the strong skeleton structure,CaO was separately fixed in certain areas,which effectively prevented the contact of more CaCO3and channel integration by CaCO3melting.Then,the porous structure was maintained.The clear pore distribution on the surface particles provided channels for CO2to spread into the cores of particles,which helped to improve the cycle stability.Different from CaO–CaAc2with high initial carbonation conversion and poor cyclic stability,CaO–Al performed stable carbonation conversion through 110 carbonation/calcination cycles,which was more looking forward to using in more cycles.
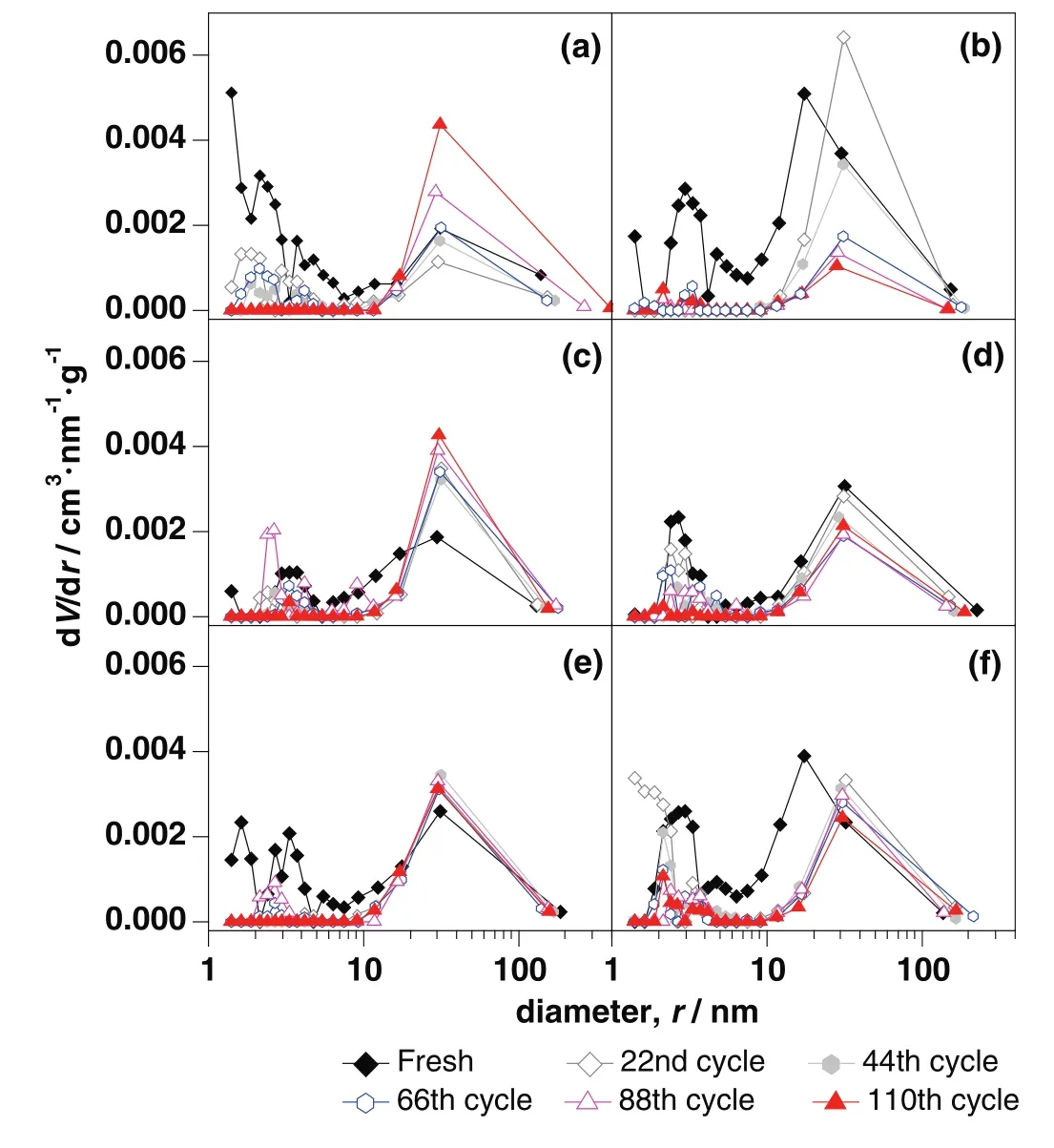
Fig.9.Pore size distribution of modified sorbents through different carbonation/calcinations:(a)CaO–CaAc2,(b)CaO–Mg,(c)CaO–Al,(d)CaO–Ce,(e)CaO–Zr,(f)CaO–La.
4.Conclusions
In present study,the CaO-based sorbents with CaAc2as precursor were modified by respectively doping Mg,Al,Ce,Zr and La,so as to improve the cyclic stabilities of the sorbents.
During the 40 min carbonation of fresh sorbents,the carbonation conversions of sorbents doped with La,Mg and Ce were close to that of CaO–CaAc2.The carbonation conversion of sorbent doped with Al or Zr was much lower than that of CaO–CaAc2due to the sacrifice of effective CaO,which reacts with Al or Zr to generate Ca12Al14O33or CaZrO3.
Through 22 carbonation/calcinations,the cyclic stabilities of all modified sorbents were much better than that of CaO–CaAc2.The cyclic stabilities of the sorbents doped with Al,Ce and La were above 96.2%after 22 cycles.
For the extended cycles(110 cycles),the decay of carbonation conversion for the modified sorbents was different,such as from sharp to slow(doped with Mg or Zr),from slow to sharp(doped with La),and at almost uniform rate(doped with Al or Ce).However,the cyclic stability for all the modified sorbents through 110 cycles was much higher than that of CaO–CaAc2,especially for doping Al,Ce and Mg.
With the almost constant critical time and stable skeleton structure made by inert Ca12Al14O33,the cyclic stability of CaO–Al was as high as 87.1%after 110 cycles.Due to the significantly cyclic stability and relatively low price,CaO–Al should be a potential CO2sorbent at high temperature for industrial application.

Fig.10.SEM of sorbents after 110 carbonation/calcination cycles:(a)CaO–CaAc2,(b)CaO–Mg,(c)CaO–Al,(d)CaO–Ce,(e)CaO–Zr,(f)CaO–La.
Nomenclature
m0sample mass before carbonation,mg
tccritical time,min
tfactual time of rapid reaction control stage,min
trretardation time,min
SNcyclic stability throughNcycles
Xcarbonation conversion
α coefficient of effective mass
[1]H.Lu,E.P.Reddy,P.G.Smirniotis,Calcium oxide based sorbents for capture of carbon dioxide at high temperatures,Ind.Eng.Chem.Res.45(2006)3944–3949.
[2]M.V.Iyer,H.Gupta,B.B.Sakadjian,L.-S.Fan,Multicyclic study on the simultaneous carbonation and sulfation of high-reactivity CaO,Ind.Eng.Chem.Res.43(2004)3939–3949.
[3]B.T.Zhang,M.Fan,A.E.Bland,CO2separation by a new solid K–Fe sorbent,Energy Fuel25(2011)1919–1925.
[4]B.Dutcher,M.Fan,B.Leonard,Use of multifunctional nanoporous TiO(OH)2for catalytic NaHCO3decomposition-eventually for Na2CO3/NaHCO3based CO2separation technology,Sep.Purif.Technol.80(2011)364–374.
[5]B.Dutcher,M.Fan,B.Leonard,M.D.Dyar,J.Tang,E.A.Speicher,P.Liu,Y.Zhang,Use of nanoporous FeOOH as a catalytic support for NaHCO3decomposition aimed at reduction of energy requirement of Na2CO3/NaHCO3based CO2separation technology,J.Phys.Chem.C115(2011)15532–15544.
[6]A.Tuwati,M.Fan,A.G.Russell,J.Wang,H.F.M.Dacosta,New CO2sorbent synthesized with nanoporous TiO(OH)2and K2CO3,Energy Fuel27(2013)7628–7636.
[7]D.Karami,N.Mahinpey,Highly active CaO-based sorbents for CO2capture using the precipitation method,preparation and characterization of the sorbent powder,Ind.Eng.Chem.Res.51(2012)4567–4572.
[8]B.W.Wang,X.Y.Song,Z.H.Wang,C.G.Zheng,Preparation and application of the sol––gel combustion synthesis-made CaO/CaZrO3sorbent for cyclic CO2capture through the severe calcination condition,Chin.J.Chem.Eng.22(2014)991–999.
[9]D.Alvarez,M.Pena,A.G.Borrego,Behavior of different calcium-based sorbents in a calcination/carbonation cycle for CO2capture,Energy Fuel21(2007)1534–1542.
[10]J.Blamey,M.Zhao,V.Manovic,E.J.Anthony,D.R.Dugwell,P.S.Fennell,A shrinking core model for steam hydration of CaO-based sorbents cycled for CO2capture,Chem.Eng.J.291(2016)298–305.
[11]H.Dieter,A.R.Bidwe,G.Varela-Duelli,A.Charitos,C.Hawthorne,G.Scheffknecht,Development of the calcium looping CO2capture technology from lab to pilot scale at IFK,University of Stuttgart,Fuel127(2014)23–27.
[12]J.Ströhle,M.Junk,J.Kremer,A.Galloy,B.Epple,Carbonate looping experiments in a 1 MWthpilot plant and model validation,Fuel127(2014)13–22.
[13]A.Sánchez-Biezma,J.Paniagua,L.Diaz,M.Lorenzo,J.Alvarez,D.Martínez,B.Arias,M.E.Diego,J.C.Abanades,Testing postcombustion CO2capture with CaO in a 1.7 MWtpilot facility,Energy Procedia37(2013)1–8.
[14]M.J.Al-Jeboori,M.Nguyen,C.Dean,P.S.Fennell,Improvement of limestone-based CO2sorbents for Ca looping by HBr and other mineral acids,Ind.Eng.Chem.Res.52(2013)1426–1433.
[15]R.Y.Sun,Y.J.Li,S.M.Wu,C.T.Liu,H.L.Liu,C.M.Lu,Enhancement of CO2capture capacity by modifying limestone with propionic acid,Powder Technol.233(2013)8–14.
[16]Z.S.Li,N.S.Cai,Y.Y.Huang,Effect of preparation temperature on cyclic CO2capture and multiple carbonation–calcination cycles for a new Ca-based CO2sorbent,Ind.Eng.Chem.Res.45(2006)1911–1917.
[17]R.Wu,S.F.Wu,Performance of nano-CaCO3coated with SiO2on CO2adsorption at high temperature,J.Chem.Ind.Eng.(China)57(7)(2006)1722–1726.(in Chinese)
[18]R.Y.Sun,Y.J.Li,H.L.Liu,S.M.Wu,C.M.Lu,CO2capture performance of calcium-based sorbent doped with manganese salts during calcium looping cycle,Appl.Energy89(2012)368–373.
[19]L.Vieille,A.Govin,P.Grosseau,Improvements of calcium oxide based sorbents for multiple CO2capture cycles,Powder Technol.228(2012)319–323.
[20]T.He,B.Cao,J.Y.Hu,J.C.He,X.Mu,L.Xu,Y.Liu,X.X.Ma,Research on calcium oxidebased sorbents for adsorption of CO2at high temperature,J.Chem.Ind.Eng.(China)35(2007)8–11.(in Chinese)
[21]X.T.Liu,X.X.Ma,Modified calcium-based sorbent for CO2capture at high temperature,The 12th Japan–China Symposium on Coal and C1 Chemistry,Fukuoka,Japan,2013.
[22]X.T.Liu,X.X.Ma,J.F.Shi,The effect of doping CeO2for CO2capture of calcium-based sorbent at high temperature,30th Annual International Pittsburgh Coal Conference,Beijing,China,2013.
[23]X.T.Liu,X.X.Ma,S.S.Xu,C.Li,D.Lu,Modified calcium-based sorbent doped with CeO2for CO2capture at high temperature,The 7th International Conference on Separation Science and Technology,Chengdu,China,2013.
[24]C.Ma,The Study of Modified Calcium-base Sorbent for CO2at High Temperature,M.S.Thesis,Northwest Univ.,China,2010.
[25]S.P.Wang,H.Shen,S.S.Fan,Y.J.Zhao,X.B.Ma,J.L.Gong,CaO-based meshed hollow spheres for CO2capture,Chem.Eng.Sci.135(2015)532–539.
[26]S.P.Wang,H.Shen,S.S.Fan,Y.J.Zhao,X.B.Ma,J.L.Gong,Enhanced CO2adsorption capacity and stability using CaO-based adsorbents treated by hydration,AIChE J.59(2013)3586–3593.
[27]S.F.Cui,W.S.Yang,Z.N.Qian,Research thermal decomposition of lanthanum hydroxide by thermogravimetry,Chem.J.Chin.Univ.8(3)(1987)271–272.(in Chinese)
[28]C.Luo,Y.Zheng,N.Ding,Q.L.Wu,G.Bian,C.G.Zheng,Development and performance of CaO/La2O3sorbents during calcium looping cycles for CO2capture,Ind.Eng.Chem.Res.49(2010)11778–11784.
[29]M.Broda,A.M.Kierzkowska,C.R.Müller,Influence of the calcination and carbonation conditions on the CO2uptake of synthetic Ca-based CO2sorbents,Environ.Sci.Technol.46(2012)10849–10856.
[30]H.Lu,A.Khan,S.E.Pratsinis,P.G.Smirniotis,Flame-made durable doped-CaO nanosorbents for CO2capture,Energy Fuel23(2009)1093–1100.
[31]P.Q.Lan,S.F.Wu,Mechanism for self-reactivation of nano-CaO-based CO2sorbent in calcium looping,Fuel143(2015)9–15.
[32]V.Manovic,E.J.Anthony,Thermal activation of CaO-based sorbent and selfreactivation during CO2capture looping cycles,Environ.Sci.Technol.42(2008)4170–4174.
[33]S.P.Wang,S.L.Yan,X.B.Ma,J.L.Gong,Recent advances in capture of carbon dioxide using alkali-metal-based oxides,Energy Environ.Sci.4(2011)3805–3819.
[34]G.W.Wu,C.X.Zhang,S.R.Li,Z.Q.Huang,S.L.Yan,S.P.Wang,X.B.Ma,J.L.Gong,Sorption enhanced steam reforming of ethanol on Ni–CaO–Al2O3multifunctional catalysts derived from hydrotalcite-like compounds,Energy Environ.Sci.5(2012)8942–8949.
[35]D.Alvarez,J.C.Abanades,Determination of the critical product layer thickness in the reaction of CaO with CO2,Ind.Eng.Chem.Res.44(2005)5608–5615.
[36]Z.S.Li,F.Fang,X.Y.Tang,N.S.Cai,Effect of temperature on the carbonation reaction of CaO with CO2,Energy Fuel26(2012)2473–2482.
[37]Z.S.Li,N.S.Cai,Y.Y.Huang,H.J.Han,Synthesis,experimental studies,and analysis of a new calcium-based carbon dioxide absorbent,Energy Fuel19(2005)1447–1452.
[38]Q.Shi,S.F.Wu,M.Z.Jiang,Q.H.Li,Reactive sorption–decomposition kinetics of nano Ca-based CO2sorbents,CIESC J.60(3)(2009)641–648.(in Chinese)
[39]A.Akgsornpeak,T.Witoon,T.Mungcharoen,J.Limtrakul,Development of synthetic CaOsorbents via CTAB-assisted sol–gel method for CO2capture athigh temperature,Chem.Eng.J.237(2014)189–198.
[40]L.Y.Li,D.L.King,Z.M.Nie,C.Howard,Magnesia-stabilized calcium oxide absorbents with improved durability for high temperature CO2capture,Ind.Eng.Chem.Res.48(2009)10604–10613.
猜你喜欢
装备维修技术(2021年36期)2021-10-25
弹箭与制导学报(2021年3期)2021-07-30
中国科技纵横(2020年16期)2021-01-27
网印工业(2019年4期)2019-05-21
重型机械(2019年2期)2019-04-28
吉首大学学报(自然科学版)(2018年3期)2018-07-03
化工学报(2017年11期)2017-11-22
工业设计(2016年7期)2016-05-04
制造业自动化(2015年18期)2015-04-25
浙江师范大学学报(自然科学版)(2012年2期)2012-12-17
 Chinese Journal of Chemical Engineering2017年5期
Chinese Journal of Chemical Engineering2017年5期
- Chinese Journal of Chemical Engineering的其它文章
- Influence of Na+,K+,Mg2+,Ca2+,and Fe3+on filterability and settleability of drilling sludge☆
- An optimal filter based MPC for systems with arbitrary disturbances☆
- Measurement and calculation of solubility of quinine in supercritical carbon dioxide☆
- Solubility and metastable zone width measurement of 3,4-bis(3-nitrofurazan-4-yl)furoxan(DNTF)in ethanol+water
- Partition coefficient prediction of Baker's yeast invertase in aqueous two phase systems using hybrid group method data handling neural network
- The effect of transition metal ions(M2+=Mn2+,Ni2+,Co2+,Cu2+)on the chemical synthesis polyaniline as counter electrodes in dye-sensitized solar cells☆