
cblC型甲基丙二酸血症合并同型半胱氨酸血症临床资料分析
2017-09-15 16:03,,
中国妇幼健康研究 2017年9期
,,
(西安交通大学医学院第一附属医院,陕西 西安710061)
cblC型甲基丙二酸血症合并同型半胱氨酸血症临床资料分析
陈萧群,周熙惠,雷玲霞
(西安交通大学医学院第一附属医院,陕西西安710061)
目的分析cblC型甲基丙二酸血症合并同型半胱氨酸血症(cblC病)患者的临床、生化特点及基因突变特点,以提高临床医生对该病近年来的临床特点的认识。方法回顾性分析2014至2016年5例cblC病患者的临床资料,总结临床表现、辅助化验检查和基因突变的特点,并对其临床特征及转归情况进行总结。结果临床特点:5例患儿起病在婴儿期,均存在有不同程度的喂养困难、精神智力运动发育落后、营养不良;血C3(丙酰肉碱)、C3/C2(丙酰肉碱与乙酰肉碱比值)、C3/C0(丙酰肉碱与游离肉碱比值)、尿甲基丙二酸、乳酸及血同型半胱氨酸均增高,C3/C2和C3/C0比C3增高更显著,3例患儿血氨轻度增高,4例头颅MRI异常。基因突变特点:共发现6个基因突变,c.104T>G(p.L35W) 、c.482G>A(p.R161Q)、c.567_568insT(p.I190fs)、656_658del(p.219_220del)及217C>T(p.R73X)各1例;c.440-441del(p.G147fs)2例;c.609G>A(p.W203X)3例,609G>A是本研究中突变最多的类型。临床转归:给予VitB12、叶酸、左旋肉碱、甜菜碱及VitB6等治疗后病情明显好转。结论cblC病的主要临床特征包括有不同程度的喂养困难、精神智力运动发育落后、营养不良;C3/C0、C3/C2、尿甲基丙二酸和同型半胱氨酸明显增高,C3/C2和C3/C0比C3增高更显著;MMACHC基因的突变是cblC病的原因;了解cblC病在婴儿期的各种临床表现和新生儿血串联质谱检测的重要性,可使此类疾病得到及时的诊断和干预,减少此类病人的致残率。
cblC型甲基丙二酸血症;临床特点;基因突变;临床转归
cblC型甲基丙二酸血症合并同型半胱氨酸血症(cblC病)是一种先天性胞内钴胺素代谢异常、临床表现多样的常染色体隐性遗传疾病,是最常见的钴胺素代谢性的遗传性疾病。此类代谢疾病是因MMACHC基因突变导致的细胞内腺苷钴胺素和甲基钴胺素合成不足,从而使相应的甲硫氨酸合成酶和甲基丙二酰辅酶A变位酶活性降低,体内甲基丙二酸、同型半胱氨酸蓄积及蛋氨酸产生减少是这种疾病的生化特征。临床表现上缺乏特异性,主要表现为不同程度的喂养困难、精神智力运动发育落后、营养不良等。不同年龄患者临床表现各异,诊断困难,随着病程增加亦可导致多系统损害[1]。cblC病多于婴幼儿期发病,我国报道的有详细的临床资料并行MMACHC基因检测的甚少,现对5例患儿的临床与实验室进行回顾性分析,旨在探讨本症近年来的诊治情况。
1对象与方法
1.1研究对象
2014至2016年西安交通大学医学院第一附属医院儿科发现cblC病患儿5例,男1例,女4例;分别来自青岛、陕西;起病年龄为2个月21天~1岁1月,病程1个月~1年;就诊年龄为2个月21天~1岁5月,均无异常家族史。
1.2研究方法
全部患儿先后行常规生化检测、脑MRI扫描、血串联质谱(MS-MS)、气相色谱-质谱(GC-MS)及MMACHC基因测序,1例进行了脑电图检测,并进行门诊复诊或电话随诊的方式进行了3~6个月的随访。
1.3统计学方法
采用SPSS 18.0统计软件进行统计学分析。对5例cblC病患儿的临床资料进行回归分析和影响因素分析总结。
2结果
2.1临床表现及头颅MRI检查
5例患儿均在婴儿期慢性起病,有不同程度的体重增长缓慢、精神智力运动发育落后、四肢肌张力减低和喂养困难。3例皮肤弹性差、营养差;反应迟钝1例;活动少1例;夜间规律性哭吵伴发热1例;男3例,女2例;4例行头颅MRI显示不同程度的脑损害,见表1。

表1 患儿临床表现、头颅MRI检查结果
2.2辅助检查
2.2.1 实验室检测
5例患儿实验室检验情况见表2。5例患儿C3/C2、C3/C0、尿甲基丙二酸、血同型半胱氨酸均增高,尿液甲基丙二酸(正常值0.2~3.6μmol/L)波动在196~493nmol/mg之间,C3/C2(正常值<0.4)、C3/C0(正常值<0.3)显著增髙。血C3明显增高4例,病例1治疗前后的MS-MS和GS-MS结果见图1(氨基酸扫描图谱)、图2(肉碱扫描图谱)、图3(尿有机酸扫描图谱),治疗后均基本恢复正常。5例患儿的血浆同型半胱氨酸50~85μmol/L(正常对照5~15μmol/L)均明显增高;甘氨酸、叶酸、肝肾功、心肌酶、甲功及苯丙氨酸正常;不同程度的酸中毒,乳酸轻度增高;1例低血糖;4例血红蛋白轻度降低;3例血氨轻度增高。

表2 初次生化检测、血MS/MS和尿GS-MS结果

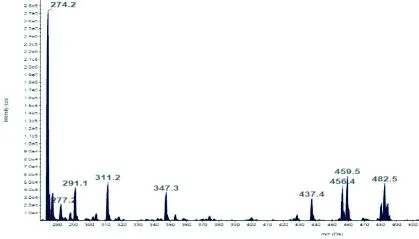
a)治疗前氨基酸扫描图谱 b)治疗后氨基酸扫描图谱
图1病例1治疗3月前后氨基酸扫描图谱
Fig.1 Amino acids scan map of case 1 before and after 3 month’s treatment
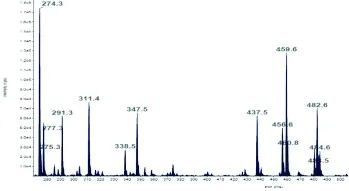

a)治疗前肉碱扫描图谱 b)治疗后肉碱扫描图谱
图2病例1治疗3月前后肉碱扫描图谱
Fig.2 Carnitine scan map of case 1 before and after 3 month’s treatment


a)治疗前尿有机酸扫描图谱 b)治疗后尿有机酸扫描图谱
图3病例1治疗3月前后尿有机酸扫描图
Fig.3 Urine organic acid scan map of case 1 before and after 3 month’s treatment
2.2.2 基因检测
5例患儿和父母均进行了MMACHC基因测序,均检测到MMACHC基因的两个杂合突变见表3,分别来自父母的基因突变,有3例为609G>A杂合突变,是突变最多的类型。图4列举了病例1的基因检测图谱。5例患儿的基因检测结果见表3。

表3 5例病例的基因检测结果

a)病例1 b)病例1之父 c)病例1之母 d)病例1 e)病例1之父 f)病例1之母

(NG16070393-1),chr1:45973050存在c.104>G杂合突变(NG16070393-1),chr1:45973050存在c.104>G杂合突变(SH16080567-1),chr1:45973050无突变(NG16070393-2),chr1:45974478存在c.440_441del的杂合突变(SH16080566-2),chr1:45974478无突变(SH16080567-2),chr1:45974478存在c.440_441del的杂合突变
图4病例1基因受检位点及sanger测序图片
Fig. 4 Gene locus tested and sanger sequencing of case 1
2.3治疗与预后
饮食方面给予普通正常饮食,5例患儿初期药物治疗为①VitB12:肌内注射0.3mg·kg-1·d-1,每日1次,共7~10天;②左旋肉碱:一般为50~100mg·kg-1·d-1;③甜菜碱:250mg·kg-1·d-1,分3次;④叶酸:5~15mg/d,分2~3次;⑤VitB6:12~30mg/d,并进行智力及运动功能训练,监测肝肾功能、血气分析、尿甲基丙二酸、血清同型半胱氨酸及血浆肉碱等指标3~6个月均恢复正常,临床症状明显改善,智力运动发育情况良好。
3讨论
3.1 cblC病的分型及发病机制
cblC病是一种先天性胞内VitB12代谢异常所导致的疾病。此类代谢疾病是位于染色体1p34.1的MMACHC基因的突变引起的疾病,MMACHC突变可导致细胞内腺苷钴胺素和甲基钻胺素合成不足引起相应的甲硫氨酸合成酶和甲基丙二酰辅酶A变位酶活性降低,从而使体内甲基丙二酸、同型半胱氨酸蓄积及蛋氨酸产生减少,可以导致多系统损害的症状,主要影响眼睛和大脑[1]。根据症状出现的早晚,临床上分为早发型和晚发型:早发型通常在出生后的第一年就表现出严重和多器官损害的症状,而晚发症状较轻,主要表现为神经系统,轻型患者可能终生不发病。cblC病的临床表型与基因型有明确的相关性,根据mRNA稳定性和蛋白质残留功能可预测疾病严重程度,这种分型的分子基础是不太清楚的[2]。 MMACHC基因的检测为cblC病精准诊断提供了分子学可靠依据。有超过100个已知突变确定导致cblC病MMACHC基因疾病,Lerner-Ellis等(2009年)报道有超过95%患者可以被测到基因突变位点,这些部分可能包括通过分析基因缺失或重复检测到缺失的外显子。许多早发型的患者,临床表现重的有c.271dupA或c.331C>T(p.R111X)突变纯合子或复合杂合子,通常迟发型的突变包括NM_015506、c.392_394del[3]和c.394C>T(p.R132X)、c.482G>A(p.R161Q)[2]。本研究5例MMACHC基因均有两个杂合突变,发现突变的类型有:c.104T>G(p.L35W)、c.482G>A(p.R161Q)、c.567_568insT(p.I190fs)、656_658del(p.219_220del)及217C>T(p.R73X)各1例;c.440-441del(p.G147fs)2例;c.609G>A(p.W203X)3例,609G>A是本研究中突变最多的类型,5例患儿临床症状相对较轻,发病隐匿,呈慢性进行性加重过程,可能与5例患者的基因检测均为杂合突变及未测出c.271dupA或c.331C>T(p.R111X)突变有关。
3.2 cblC病的临床特点及诊断
cblC病诊断主要根据临床表现、常规检验、MS-MS初筛、GS-MS复查及MMACHC基因突变检测。本研究中5例均有不同程度的喂养困难、精神智力运动发育落后、营养不良,合并双侧隐睾、卵圆孔未闭、动脉导管未闭及脐疝各1例,这些因素与本病无关,均为混杂因素。从患儿的胎龄及体重看出此病不会导致早产和明显的宫内体格发育异常。在临床实践中,对慢性起病、喂养困难、智力运动发育落后或倒退、不明原因的哭吵、脱水、贫血等为主要临床表现的患儿,首先进行常规生化检测(血尿常规、血气分析、血糖、血氨、肝肾功能、心肌酶及同型半胱氨酸)和头颅MRI排除常见疾病情况下,有代谢性酸中毒伴阴离子间隙增高、高氨血症、血液系统改变的患儿,尽早采用“MS-MS初筛、GS-MS复查”方式。MS-MS可快速检测cblC病患儿血液中甲基丙二酸和C3增高,而且C3/C0和C3/C2显著升高。对于部分在急性期可能因呕吐或食欲差的患儿,因肉碱摄入减少,血液中C3可能是正常的,但其他酰基肉碱因摄入肉碱减少而降低,但C3/C0和C3/C2仍是显著升高的;也有cblC病的新生儿筛查提示低肉碱和C3正常的报道[4],故C3/C0和C3/C2对cblC病的诊断比C3增高更有价值。本研究中4例血甲基丙二酸和C3增高,1例不显著,但C3/C2、C3/C0均显著增高。GS-MS能快速精确检测cblC病患儿尿中甲基丙二酸是明显增高的,由于其他疾病发生酸中毒时尿中甲基丙二酸也会轻度增高,故GS-MS应结合MS-MS对本病进行初步诊断,由于GS-MS重复性高,饮食、补液、纠酸等因素对检测结果均无影响,二者结合还能快速评估cblC病的治疗监测及疗效评估[5]。在cblC病患儿中血浆同型半胱氨酸是升高的,血氨有不同程度的增高,但一般不会超过100μmol/L,这也是区别于尿素循环障碍所至的高氨血症。本研究中5例尿甲基丙二酸、血同型半胱氨酸均增高,血氨只有3例是轻度增高的。对于C3、C3/C2、C3/C0、尿甲基丙二酸及血同型半胱氨酸增高超过界限值,怀疑cblC病的患儿需行MMACHC基因突变检测才能确诊。
此外,cblC病需与继发性cblC病、丙酸血症进行鉴别。由于VitB12测定可在基因检测前进行,故血VitB12测定可鉴别原发性与继发性cblC病。本研究中5例的VitB12水平均正常。继发性的如因先天性VitB12吸收障碍、摄入不足所致的巨幼红细胞性贫血的患儿,血VitB12水平测定是明显降低的[6]。丙酸血症的患儿除有C3、C3/C0及C3/C2升高外,常伴有甘氨酸显著升高[4],且尿中3-羟基丙酸及甲基枸橼酸升高显著,但甲基丙二酸水平正常。在本次研究中5例的甘氨酸均正常。
由于cblC病起病隐匿,容易漏诊和误诊,故新生儿出生1周内采血行MS-MS初筛是早期诊断的有效方法。cblC病的早期遗传学产前诊断[7]是对高危孕妇进行羊水胎儿细胞或绒毛膜绒毛取样进行MMACHC基因突变检测,Huemer等(2005年)证实cblC病胎儿的代谢物可以在孕产妇羊水和尿液中被检测和监测。目前我国对cblC病患儿母亲再次妊娠16~20周时抽取羊水进行GC-MS、MS-MS和MMACHC基因突变检测来确诊。
3.3治疗
急性期治疗的目标是改善临床症状,尽快纠正血清蛋氨酸和降低血浆半胱氨酸和甲基丙二酸水平。除了应给予足够的能量摄入,若存在体液和基础能量失衡需及时纠正和治疗。一旦怀疑cblC病时首先尽快给予VitB12和甜菜碱治疗,而不必等待检测血浆代谢物和VitB12水平达标后再给予治疗,这样可以显著改善临床症状、代谢产物和生长发育,另外给予左卡尼汀可以促进丙酰族物质的排泄,预防肉碱缺乏症[8];补充叶酸和VitB6作为cblC病患者的辅助治疗。
cblC病患儿的预后取决于MMACHC基因突变的分型、发现早晚和长期规律治疗三方面。早诊早治可有效改善预后,产前的早期给予高剂量VitB12治疗可以预防出现cblC病患儿的临床症状[9]。
综上所述,近年来,cblC病越来越受到医学界的重视,对其发生机制的研究,尤其是分子生物学研究的进展,使儿科医生对此疾病的认识进一步深化,使不少患儿在早期就得到了及时的临床诊断和治疗。本文通过5例患儿MS-MS、GC-MS及MMACHC基因突变检测明确诊断cblC病临床资料进行分析总结,使我们对本病的早期识别有了意识,减少漏诊误诊,对高危胎儿的筛检和扩大新生儿筛查的覆盖率,可减少婴幼儿的病死率及致残率。随着临床医生对cblC病的认识和警惕,对于确诊cblC病的患儿进行系统的管理,原有迟发型所占比例会下降。
[1]Fuchs L R,Robert M,Ingster-Moati I,etal.Ocular manifestations of cobalamin C type methylmalonic aciduria with homocystinuria[J].J AAPOS, 2012, 16(4):370-375.
[2]Martinelli D,Deodato F,Dionisi-Vici C.Cobalamin C defect: natural history, pathophysiology, and treatment[J].J Inherit Metab Dis,2011,34(1):127-135.
[3]Backe P H,Ytre-Arne M,Røhr A K,etal.Novel deletion mutation identified in a patient with late-onset combined methylmalonic acidemia and homocystinuria, cblC type[J].JIMD Rep,2013,11:79-85.
[4]Ahrens-Nicklas R C,Serdaroglu E,Muraresku C,etal.Cobalamin C disease missed by newborn screening in a patient with low carnitine level[J].JIMD Rep,2015,23:71-75.
[5] 杨楠,韩连书,叶军,等.新生儿期氨基酸、有机酸及脂肪酸氧化代谢病疾病谱分析[J].临床儿科杂志,2012,30(9):805-808.
[6] 刘小梅,陈植,袁林,等.以肾脏损害为首发症状的儿童甲基丙二酸尿症9例分析[J].中国循证儿科杂志,2010,5(6):447-451.
[7]Zong Y,Liu N,Zhao Z,etal.Prenatal diagnosis using genetic sequencing and identification of a novel mutation in MMACHC[J].BMC Med Genet,2015,16:48.
[8]Carrillo-Carrasco N,Chandler R J,Venditti C P.Combined methylmalonic acidemia and homocystinuria, cblC type. I. Clinical presentations, diagnosis and management[J].J Inherit Metab Dis,2012,35(1):91-102.
[9]Trefz F K,Scheible D,Frauendienst-Egger G,etal.Successful intrauterine treatment of a patient with cobalamin C defect[J].Mol Genet Metab Rep,2016,6:55-59.
[专业责任编辑:艾 婷]
[妇幼营养研究]
Clinical data analysis of cblC methylmalonic acidemia combined with homocysteinemia
CHEN Xiao-qun, ZHOU Xi-hui, LEI Ling-xia
(FirstAffiliatedHospitalofXi’anJiaotongUniversity,ShaanxiXi’an710061,China)
ObjectiveTo analyze the clinical, biochemical and gene mutation features in patients with cblC methylmalonic acidemia combined with homocysteinemia (cb1C disease) and to improve knowledge of clinicians on clinical features of this disease in recent years.MethodsClinical data of 5 patients diagnosed with cblC disease from 2014 to 2016 was retrospectively analyzed. Clinical manifestations, laboratory examinations and gene mutation features were summarized and their clinical features and prognosis were summarized too.ResultsClinical features were as following: onset of disease of all five patients was in infancy, who manifested various degree of feeding difficulty, development backwardness of mental, intelligence and sport ability, malnutrition. Levels of C3 (propionyl-carnitine), C3/C2 (ratio of propionyl-carnitine and acetylcamitine), C3/C0 (ratio of propionyl-carnitine and free carnitine), urine methylmalonic acid, lactic acid and serum homocysteine were increased. Increases of C3/C2 and C3/C0 were more significant than increase of C3. Blood ammonia increased slightly in 3 cases and head MRI scanning results of 4 patients were abnormal. Gene mutation features showed that 6 gene mutations were found, including c.104T>G (p.L35W), c.482G>A (p.R161Q), c.567_568insT (p.I190fs), 656_658del (p.219_220del) and 217C>T(p.R73X) with each mutation presented in one case, c.440_441del (p.G147fs) in 2 cases, c.609G>A(p.W203X) in 3 cases, and 609G>A was the most common mutation type found in this study. Clinical outcome was that all patients improved evidently after treatment of vitamin B12, folic acid, L-camitine, betaine, and vitamin B6.ConclusionThe main clinical features of cblC disease include various degree of feeding difficulty, development backwardness of mental, intelligence and sport ability, malnutrition, significant increase of C3/C0, C3/C2, urine methylmalonic acid and homocysteine, and more significant increase of C3/C2 and C3/C0 than C3. Gene mutation of MMACHC is the cause of cblC disease. Knowledge on various clinical manifestations of cb1C disease in infancy and awareness of importance of neonatal blood tandem mass spectrometry allows early detection and interference of this disease and reduce disability rate of patients.
cblC methylmalonic acidemia combined with homocysteinemia (cblC disease); clinical features; gene mutations; clinical outcome
2017-04-05
陈萧群(1974—),女,副主任医师,主要从事儿童内分泌的研究。
10.3969/j.issn.1673-5293.2017.09.031
R725
A
1673-5293(2017)09-1133-04
猜你喜欢
昆明医科大学学报(2021年1期)2021-02-07
中国生殖健康(2020年2期)2021-01-18
中华养生保健(2020年5期)2020-11-16
中国畜牧杂志(2019年4期)2019-04-20
小学生导刊(2018年13期)2018-06-29
中国生殖健康(2018年2期)2018-01-12
中国医药指南(2017年3期)2017-11-13
食品界(2017年4期)2017-05-17
家庭百事通·健康一点通(2017年3期)2017-03-22
医学研究杂志(2015年12期)2015-06-10
