
聚酰亚胺/功能化石墨烯复合材料力学性能及玻璃化转变温度的分子动力学模拟∗
2017-12-05 02:35杨文龙韩浚生王宇林家齐2何国强孙洪国
物理学报 2017年22期
杨文龙 韩浚生 王宇 林家齐2) 何国强 孙洪国
1)(哈尔滨理工大学应用科学学院,哈尔滨 150080)2)(哈尔滨理工大学,工程电介质及其应用教育部重点实验室,哈尔滨 150080)3)(广西大学材料科学与工程学院,有色金属及特色材料加工国家重点实验室培养基地,南宁 530004)4)(中国科学院长春应用化学研究所高分子复合材料工程实验室,长春 130022)
聚酰亚胺/功能化石墨烯复合材料力学性能及玻璃化转变温度的分子动力学模拟∗
杨文龙1)†韩浚生1)王宇1)林家齐1)2)何国强3)孙洪国4)
1)(哈尔滨理工大学应用科学学院,哈尔滨 150080)2)(哈尔滨理工大学,工程电介质及其应用教育部重点实验室,哈尔滨 150080)3)(广西大学材料科学与工程学院,有色金属及特色材料加工国家重点实验室培养基地,南宁 530004)4)(中国科学院长春应用化学研究所高分子复合材料工程实验室,长春 130022)
(2017年5月20日收到;2017年8月20日收到修改稿)
应用分子模拟方法,建立了聚酰亚胺(polyimide,PI),石墨烯及羧基、氨基、羟基功能化石墨烯模型,探究了聚酰亚胺和石墨烯,聚酰亚胺和功能化石墨烯共混后复合材料的力学性能和玻璃化转变温度(Tg).研究结果表明,羧基修饰的石墨烯与PI复合后材料力学性能增加显著,其杨氏模量和剪切模量分别为4.946 GPa和1.816 GPa.不同官能团修饰的石墨烯引入PI后材料的Tg均有不同程度下降;未修饰的石墨烯与PI复合后,其Tg(559.30 K)较纯PI的Tg(663.57 K)降幅最大;而羧基修饰的石墨烯与PI复合后Tg(601.61 K)降幅最小.计算比较了PI/石墨烯复合材料体系密度、溶解度参数、相互作用能、弹性系数和氢键平均密度,研究发现羧基修饰石墨烯/PI复合材料的密度为1.396 g.cm−3,溶解度参数为23.51 J1/2.cm−3/2,其相互作用能与氢键平均密度最大,弹性系数显示羧基修饰石墨烯与PI组成的复合材料内部最均匀.计算结果表明,羧基功能化石墨烯可以大幅度提高PI的力学性能,增强石墨烯与PI之间的相互作用可以减少复合材料Tg的降幅程度.此基体间相互作用的研究方法可以作为预测聚合物基纳米复合材料结构与性能的有效工具,以期为材料的设计与应用提供理论指导.
聚酰亚胺,石墨烯,分子动力学模拟,玻璃化转变温度
1 引 言
聚合物基纳米复合材料是一种集聚合物和纳米填料优点于一身的结构或功能材料,如高强度、轻质、耐磨、低介电等.因此,通过纳米填料修饰聚合物,设计并合成满足不同实际需求的复合材料成为材料领域的研究热点[1,2].石墨烯(graphene,G)是一种碳原子以sp2杂化链接的单原子层石墨,这种独特的二维结构赋予石墨烯优异的力学、热学、电学和光学等性能,如其杨氏模量接近1 TPa,断裂强度高达130 GPa,断裂伸长率约25%,禁带宽度几乎为零,热导率可达到5000 J.m−1K−1.s−1、宽频的光吸收和光学非线性等[3−5].这些优异的性能使得石墨烯及其复合材料可以在超级电容器、太阳能电池、纳米器件等诸多领域中都有着广泛的应用前景.
在石墨烯复合材料中,石墨烯与聚合物之间存在较弱的界面相互作用限制了其实际合成及应用[6].研究表明,对石墨烯的表面改性是一种提高石墨烯复合材料中界面强度的有效途径,而且对于不同的聚合物性能有显著提高[7−16].适量的表面改性石墨烯和聚乙烯(polyethylene,PE)[17]、聚甲基丙烯酸甲酯[18]、聚乙烯醇[19]、聚氯乙烯[20]复合,能够显著提高其断裂强度和断裂伸长率;氧化石墨烯(graphene oxide,GO)可以大幅度增强聚酰亚胺(polyimide,PI)的机械性能,同时还可以使其相对介电常数从3.0降低到2.0附近[21];苯胺修饰的石墨烯可以将PI的电导率升高至6.6×10−2S/m,是纯PI的1011倍[22].然而在石墨烯复合材料制备和表征过程中,由于石墨烯的形态和表面结构以及诸多变量(实验温度、压力、聚集态等制约等因素)的影响,研究者难以对所报道的数据进行平行比较、数据分析,难以建立结构与材料性能之间的关系,难以确定影响材料性能的关键因素,因此对复合材料了解深度有限.
分子模拟是一种可以设定系统的各种变量(温度、压强等),详述体系微观结构并能对实验的现象进行完善说明的研究方法[23−27].分子模拟不仅仅能通过分子、原子角度对微观结构设计,还可以从定性和定量两个角度对于复合材料的结构与性能之间的关系进行解释说明.相比于传统的试验方法,分子模拟可以提供理想的试验环境,精确控制聚合物和纳米填充材料的界面结合情况,这可以为新型功能材料的设计和合成提供理论指导.我们在前期工作中采用分子动力学模拟了在聚酰亚胺中掺杂不同形态的纳米粒子,研究发现适量的纳米粒子可以显著提高聚合物基体的机械性能和热力学性能[28];Compton等[29]发现氧化石墨烯纸中含有少量水时,由于氢键等非键作用可以显著提高纸的强度;Sheng等建立了聚乙烯/石墨烯(PE/G)复合材料模型,发现随着复合体系中石墨烯的比重增加,复合材料界面之间的van der Waals作用随之减弱,导致PE/G复合材料的玻璃化转变温度(Tg)降低[30].芳香聚酰亚胺是指主链上含有芳香环、介电性能突出、化学性能稳定且在较高温度(约200◦C)仍能保证材料不失效的一类聚合物,是复合材料良好的载体[28].本文选择均苯型聚酰亚胺作为基体,选用不同官能团(羟基、羧基、氨基)修饰的石墨烯作为增强体,通过多尺度建模、利用分子模拟方法预测不同官能团修饰的石墨烯对聚酰亚胺的杨氏模量、剪切模量和玻璃化转变温度的影响,以期对石墨烯复合材料的结构与性能提供理论指导.
2 模型构建与模拟方法
2.1 模型构建
建立PI分子链和石墨烯模型.在石墨烯表面随机修饰羟基、羧基和氨基官能团,具体参数如表1.首先将PI分子链和石墨烯装箱做成一种具有代表性的周期性结构的元胞模型.然后对该模型进行几何优化,使其能量降到最低.最后进行分子动力学模拟,使其能量平稳,以便后续对模型的性能模拟计算.

表1 PI及其复合材料模型元胞组成Table 1.Cell components of PI and composite models.
2.2 分子模拟参数
羟基通常随机分布在GO的基面上,羧基通常随机分布在GO的边缘[31],氨基则可以通过GO获得[32,33].本文拟定氨基随机分布在G边缘,如图1所示.
本文利用Material Studio软件,通过Forcite模块进行分子模拟优化.分子模拟的力场为COMPASS,进行几何优化方法用smart方法,进行分子动力学优化时,压强在一个标准大气压.系综模拟时采用Andersen控温方法、Berendsen控压方法,系综的模拟步长时间均为500 ps,van der Waals和静电作用用Group based方法,力场非键截取半径取12 Å,时间步长为1 fs.进行分子动力学优化,首先是NVT(原子数恒定、体积恒定、温度恒定)系综中,温度保持298 K,使模型中分子充分松弛.然后在NPT(原子数恒定、压强恒定、温度恒定)体系中,在1个标准大气压下进行恒温、恒压压缩,使之密度更加接近于实际情况.之后再进行退火处理,进而消除装箱中的不合理构象和小概率构象.最后得到一系列的复合材料元胞,如图2所示.

图1 (网刊彩色)功能化石墨烯模型 (a)G模型;(b)GOH模型;(c)GCOOH模型;(d)GNH2模型Fig.1.(color online)Models of functional graphene:(a)G model;(b)GOH model;(c)GCOOH model;(d)GNH2model.

图2 (网刊彩色)PI/G复合物模型 (a)复合物整体图;(b)PI单体重复单元和组成原子Fig.2.(color online)PI/G composite model:(a)The model of whole composite;(b)the repeat unit and constituent atoms of PI monomer.
3 结果与讨论
3.1 复合材料结构
由于微观结构直接影响材料的性能,因此为了探究功能化石墨烯对PI力学性能和Tg的影响,需要对聚酰亚胺/功能化石墨烯复合材料的结构进行剖析.根据经典理论,物质的内聚能可以看作在零压力下将体系内所有的分子分离到无限远处所需的能量,内聚能密度是单位体积的内聚能.溶解度参数也可以表示体系内部作用力强弱,通常由内聚能密度的开方获得[28].在复合材料体系中,增强体与基体的相互作用大小也是衡量材料性能的标准之一,通常两者相互作用越强,对材料的力学性能和Tg影响越大.通过分子模拟优化后的PI及其复合材料模型参数列于表2,PI与石墨烯的相互作用列于表3.

式中,Ucoh为体系的内聚能,Uinter为所有分子间的总能量,Utotal为体系的总能量,Uinter为分子内的能量,〈···〉为体系的平均值,CED为内聚能密度,V为体系的体积,δ为溶解度参数,Uinteraction为基体与增强体的相互作用能,UPI为PI的能量,UGX为石墨烯和功能化石墨烯的能量,UPI/GX为聚酰亚胺/石墨烯复合材料的能量.
由表2可见,PI的密度1.312 g.cm−3和溶解度参数21.84 J1/2.cm−3/2, 与文献[28]中 的1.30—1.40 g.cm−3和21.17 J1/2.cm−3/2接近,说明构建优化的PI模型与实际相符.纵向对比PI及其复合材料模型的密度和溶解度参数,将石墨烯与PI复合后,材料的密度与溶解度参数均显著提高,其中PGCOOH提升最为显著,分别为1.391 g.cm−3和23.42 J1/2.cm−3/2.
由表3可见,未功能化的石墨烯与PI复合后,PG中两相相互作用能为380.99 kcal.mol−1,GCOOH与PI相互作用能在四个体系中最高,为484.81 kcal.mol−1. 通过密度、溶解度参数和相互作用能的计算对比可以发现PGCOOH与纯PI和其他三种体系相比,均具有较高的密度、溶解度参数,说明GCOOH与PI基体结合相对紧密.

表2 PI及其复合材料模型的元胞尺寸、密度和溶解度参数Table 2.Cell size,density and solubility parameters of PI and composite materials.

表3 聚酰亚胺/石墨烯复合材料中PI与石墨烯的相互作用能Table 3.Interactional energy of PI and graphene in polyimide/graphene composites.
3.2 复合材料的力学性能
力学性能是关系到材料制备、加工、生产和使用的一个重要性能.在分子模拟中,复合材料模型的力学性能经静态力学方法分析后恒应变方法计算得到.通过计算复合材料模型的内应力张量σ,然后利用Parrinello-Rahman波动方法获得由应力和应变计算得到的弹性系数矩阵Cijkl[28].

其中,下标i表示粒子标号(i=1,2,3,···,N);mi,vi,ri,fi分别表示第i个粒子的质量、速度、位置和作用在其上的力;⊗表示两个向量的并矢运算;rij表示从i原子到j原子的相对位置矢量;fij表示j原子作用在i原子上的力;V0表示体系未变形时的体积;K为玻尔兹曼常数;T是体系的温度;〈V〉是模型体积的平均值.
通常情况下,弹性系数可以表示材料对于外力的一种反应能力,而且材料的力学性能均可以由其6×6的弹性系数矩阵求得.与晶体的各向异性不同,聚合物和聚合物基纳米复合材料为无定型结构,可近似看出各向同性,满足Cij=Cji.C11,C22,C33和C44,C55,C66的方差越小,说明材料越接近各向同性,对于复合材料而言,增强体和基体的相容性更好,相互作用更强[34].模量是材料在受力状态下应力与应变的比值,表征材料对在外力作用下形变的难易程度.模量越大,使材料形变的应力越大,即材料的刚性越大.表4和表5分别列出了PI及其复合材料在298 K时的弹性系数、杨氏模量(E)和剪切模量(G).
如表4所列,PI的C11,C22,C33和C44,C55,C66方差分别为0.789,0.596,说明PI的C11,C22,C33和C44,C55,C66相差不大,可以看作各向同性体.当PI与G复合后,两组方差变化为3.068和1.289,显著升高,说明PG的刚性增加,且PI与G的相容性有待提高.将G进行表面修饰羧基、羟基和氨基后,两组方差较PG体系下降,其中PGCOOH最为明显,分别为0.672和0.488.说明在石墨烯的表面修饰羧基可以有效地提高PI和石墨烯的相互作用,是材料内部更加均匀化,在石墨烯上修饰羟基和氨基的效果较羧基差些.
如表5所列,在298 K时PG的杨氏模量E和剪切模量G分别为3.625 GPa和1.296 GPa,相比于PI的3.174 GPa和1.139 GPa有所提升,表明石墨烯加入PI后可以有效地抵抗外力作用下的形变,为PI分担更多的外力作用.在石墨烯修饰官能团后,复合体系的力学性能大幅度提升,PGCOOH的最为显著,分别为4.946 GPa和1.816 GPa.结合前面复合体系的相互作用,说明羧基功能化的石墨烯结合紧密,可以有效提高复合体系的杨氏模量E和剪切模量G.
在聚合物中掺杂无机纳米粒子后,由于聚合物和纳米粒子的相互作用会使得聚合物链段的内旋转势垒增加,抑制了高分子分子链的活动,从而提高其机械性.在众多分子间相互作用(氢键、van der Waals和静电作用等)中,O—H和N—H等弱氢键作用范围约为2.20—3.20 Å[35],对材料性能影响较为显著.因此为了进一步探究材料中分子间作用力强弱,对PI及其复合材料中氢键作用进行了定量计算.在PI及其复合材料中,氢键作用主要由N—H和O—H两部分组成.而径向分布函数可以表征材料微观结构的物理量[36],其定义式为
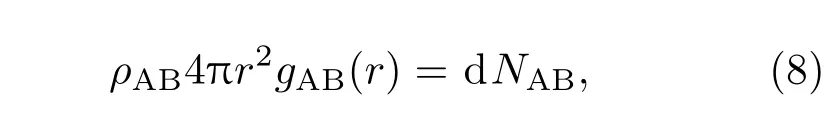
积分后得

式中,NAB为体系中A,B原子的个数;r为A和B原子之间的距离间隔宽度;ρAB为体系密度.通过(9)式即可求得体系中N(O)原子周围H原子的个数,进而求得体系内平均氢键密度,如表6.
表6列出了298 K时PI及其复合材料体系内N-H和O-H的平均氢键密度.可以发现纯PI体系中氢键密度为(2.840±0.009)×1014cm−3,在PI中填充石墨烯后使得材料内部氢键增加,PG为(3.154±0.009)×1014cm−3,PGCOOH为(3.175±0.010)×1014cm−3,PGOH 为(3.161±0.013)×1014cm−3,PGNH2为(3.171±0.011)×1014cm−3.表明纯PI修饰后,体系氢键均有所增多,两相间的相互作用越强,PI的分子链段运动时需要的能量越高,复合材料刚性增大.

表4 PI及其复合材料的弹性系数Table 4.Elastic coefficient of PI and composite materials.
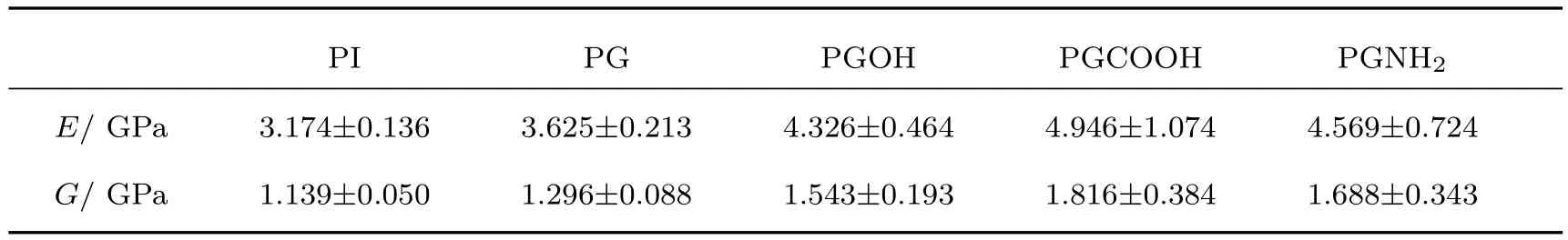
表5 PI及其复合材料的杨氏模量(E)和剪切模量(G)Table 5.Young modulus and shear modulus of PI and composite materials.

表6 PI及其复合材料中氢键平均密度Table 6.The average density of hydrogen bonds in PI and composite materials.
3.3 复合体系的Tg预测
玻璃化转变温度是聚合物的一个重要物理参量,表示高分子链段能够运动的临界温度.针对聚合物玻璃化转变的这种分子热运动现象,目前有很多理论对其进行解释.自由体积理论最先由Flory和Fox提出,该理论认为固体或者液体都是由具有原子尺寸的缺陷或由于原子无规则堆砌而产生的空穴和已经被组成物质的微粒所占据的体积构成的.这些空穴提供了原子、分子热运动的空间.在较低温度时(玻璃态),聚合物的热运动单元仅仅为分子链上的侧基或者分子键长和键角的小幅度变化,当温度达到Tg附近时,聚合物的链段具有了足够的能量,可以进行热运动,聚合物整体的自由体积陡然增加.因此,通过对材料进行不同温度下的比体积测定,可以在温度-比体积图像找到不连续变化区间,即为玻璃化转变温度[34].PI及其复合材料的玻璃化转变温度,如图3所示.

图3 PI及其复合材料的温度-比体积图 (a)PI;(b)PG;(c)PGOH;(d)PGCOOH;(e)PGNH2Fig.3.The temperature and speci fic volume of PI and composite materials:(a)PI;(b)PG;(c)PGOH;(d)PGCOOH;(e)PGNH2.
图3中通过对PI及其复合材料的温度-比体积曲线拟合计算,获得PI的Tg为663.57 K,与模拟文献[37]中400◦C相差不大.模拟复合材料的Tg中,PG的最低,为559.30 K,相比于PI下降104.27 K;PGCOOH的最高,为601.61 K,相比于PI下降61.93 K.模拟结果表明,在PI中加入G可以使其Tg降低,这主要可能由于层状的石墨烯使得PI分子链易于运动.由于GCOOH与PI的结合能和氢键密度最大,其Tg在复合体系中最高,G与PI的结合能和氢键密度最小,其Tg在复合体系中最低,表明加入适量的石墨烯可以改善聚合物的Tg.
4 结 论
建立了PI、石墨烯及羧基、氨基、羟基功能化石墨烯模型,运用分子模拟方法预测了PI/石墨烯和PI/功能化石墨烯模型的密度、溶解度参数、相互作用能、弹性系数、杨氏模量、剪切模量、Tg和氢键数目,预测修饰羧基、羟基和氨基后的石墨烯对PI力学性能和Tg的影响.通过比较材料的密度、溶解度参数和相互作用能,发现GCOOH与PI基体的相互作用比G,GOH,GNH2更强,对PI的力学性能和Tg影响更加明显.通过预测复合材料的力学性能,发现相比于PI的杨氏模量(3.174 GPa)和剪切模量(1.139 GPa),GCOOH对PI的提升最为明显,其杨氏模量和剪切模量分别为4.946 GPa和1.816 GPa.由于在石墨烯上修饰羧基后可以使复合材料内部结合紧密,相比于未修饰的石墨烯,PGCOOH更趋近于各向同性体,材料内部更加均匀.纯PI修饰后,体系氢键均有所增多,两相间的相互作用越强,PI的分子链段运动时需要的能量越高,复合材料刚性增大.通过对材料的Tg预测,表明层状的石墨烯易于PI分子链运动,其中PGCOOH的Tg(601.61 K)较纯PI(663.57 K)降幅最小,PG的Tg(559.30 K)降幅最大.综上所述,石墨烯可以显著地改善聚酰亚胺的机械性能和Tg,表面改性后的石墨烯与聚酰亚胺结合的越紧密,机械性能提高越明显,Tg下降幅度越小.
[1]Hernández M,Bernal M D M,Verdejo R,Ezquerra T A,López-Manchado M A 2012Compos.Sci.Technol.73 40
[2]Yang X,Tu Y,Li L,Shang S,Tao X M 2010ACS Appl.Mater.Inter.2 1707
[3]Gong L,Kinloch I A,Young R J,Riaz I,Jalil R,Novoselov K S 2010Physics22 2694
[4]Kuilla T,Bhadra S,Yao D,Kim N H,Bose S,Lee J H 2010Prog.Polym.Sci.35 1350
[5]Mortazavi B,Ahzi S 2013Carbon63 460
[6]Bao C,Guo Y,Song L,Kan Y,Qian X,Hu Y 2011J.Mater.Chem.21 13290
[7]Huang T,Xin Y,Li T,Nutt S,Su C,Chen H,Liu P,Lai Z 2013ACS Appl.Mater.Inter.5 4878
[8]Chen D,Zhu H,Liu T 2010ACS Appl.Mater.Inter.2 3702
[9]Huang T,Lu R,Su C,Wang H,Guo Z,Liu P,Huang Z,Chen H,Li T 2012ACS Appl.Mater.Inter.4 2699
[10]Awasthi A P,Lagoudas D C,Hammerand D C 2009Model.Simul.Mater.Sci.Eng.17 015002
[11]Boukhvalov D W,Katsnelson M I 2009J.Phys.:Condens.Matter21 344205
[12]Ha H W,Choudhury A,Kamal T,Kim D H,Park S Y 2012ACS Appl.Mater.Inter.4 4623
[13]Luong N D,Hippi U,Korhonen J T,Soininen A J,Ruokolainen J,Johansson L,S,Nam J D,Sinh L H,Seppälä J 2011Polymer52 5237
[14]Mortazavi B,Ahzi S 2013Carbon63 460
[15]Yoonessi M,Shi Y,Scheiman D A,Lebron-Colon M,Tigelaar D M,Weiss R A,Meador M A 2012ACS Nano6 7644
[16]Park O K,Kim S G,You N H,Ku B C,Hui D,Lee J H 2014Compos.Part B:Eng.56 365
[17]Kim H,Kobayashi S,AbdurRahim M A,Zhang M J,Khusainova A,Hillmyer M A,Abdala A A,Macosko C W 2011Polymer52 1837
[18]Tripathi S N,Saini P,Gupta D,Choudhary V 2013J.Mater.Sci.48 6223
[19]Liang J,Yi H,Long Z,Yan W,Ma Y,Guo T,Chen Y 2009Adv.Funct.Mater.19 2297
[20]Vadukumpully S,Paul J,Mahanta N,Valiyaveettil S 2011Carbon49 198
[21]Wang J Y,Yang S Y,Huang Y L,Tien H W,Chin W K,Ma C C M 2011J.Mater.Chem.21 13569
[22]Park O K,Hwang J Y,Goh M,Lee J H,Ku B C,You N H 2013Macromolecules46 3505
[23]Wang J,Li L,Wei Z D 2016Acta Phys.-Chim.Sin.32 321(in Chinese)[王俊,李莉,魏子栋 2016物理化学学报32 321]
[24]Hu J,Ruan X,Jiang Z,Chen Y 2009Nano Lett.9 2730
[25]Medhekar N V,Ramasubramaniam A,Ruo ffR S,Shenoy V B 2010ACS Nano4 2300
[26]Rissanou A N,Harmandaris V 2014Soft Matter10 2876
[27]Rissanou A N,Harmandaris V 2013J.Nanopart.Res.5 1
[28]Lin J Q,Li X K,Yang W L,Sun H G,Xie Z B,Xiu H J,Lei Q Q 2015Acta Phys.Sin.64 126202(in Chinese)[林家齐,李晓康,杨文龙,孙洪国,谢志滨,修翰江,雷清泉2015物理学报64 126202]
[29]Compton O C,Cranford S W,Putz K W,An Z,Brinson L C,Buehler M J,Nguyen S T 2011ACS Nano6 2008
[30]Sheng Y Z,Hua Y,Li J Y,Miao S 2013Chem.Res.Chin.U.29 788
[31]Chen J,Zhao D,Jin X,Wang C,Wang D,Ge H 2014Compos.Sci.Technol.97 41
[32]Huang T,Xin Y,Li T,Nutt S,Su C,Chen H,Liu P,Lai Z 2013ACS Appl.Mater.Inter.5 4878
[33]Zhang C,Hao R,Liao H,Hou Y 2013Nano Energy2 88
[34]Fu Y Z,Hu S Q,Lan Y H,Liu Y Q 2010Acta Chim.Sin.68 809(in Chinese)[付一政,胡双启,兰艳花,刘亚青2010化学学报68 809]
[35]Zhou G D,Duan L Y 2008Basic of Structural Chemistry(4th Ed.)(Beijing:Peking University Press)p324(in Chinese)[周公度,段连运2008结构化学基础 (第4版)(北京:北京大学出版社)第324页]
[36]Chen Z L 2007Theory and Practice of Molecular Simulation(Beijing:Chemical Industry Press)p110[陈正隆2007分子模拟的理论与实践(北京:化学工业出版社)第110—112页]
[37]Ding M X 2006Polyimide:Chemistry,Relationship between Structure and Properties and Materials(Beijing:Science Press)pp225,226(in Chinese)[丁孟贤 2006聚酰亚胺——化学、结构与性能的关系及材料(北京:科学出版社)第225,226页]
PACS:62.25.–g,71.15.Pd,81.07.NbDOI:10.7498/aps.66.227101
*Project supported by the National Natural Science Foundation of China(Grant No.61372013)and Natural Science Foundation of Heilongjiang Province,China(Grant No.E201258).
†Corresponding author.E-mail:wlyang@hrbust.edu.cn
Molecular dynamics simulation on the glass transition temperature and mechanical properties of polyimide/
functional graphene composites∗
Yang Wen-Long1)†Han Jun-Sheng1)Wang Yu1)Lin Jia-Qi1)2)He Guo-Qiang3)Sun Hong-Guo4)
1)(Department of Applied Science,Harbin University of Science Technology,Harbin 150080,China)2)(Key Laboratory of Engineering Dielectrics and Its Application,Harbin University of Science Technology,Harbin 150080,China)
3)(School of Material Science and Engineering,Cultivation Base for State Key Laboratory of Prcesing for Non-ferrous Metal and Featured Materials,Guangxi University,Nanning 530004,China)
4)(Polymer Composites Engineering Laboratory,Changchun Institute of Applied Chemistry,Chinese Academy of Sciences,Changchun 130022,China)
20 May 2017;revised manuscript
20 August 2017)
Polyimide(PI)and the functional graphene modi fied with nano-composite models of hydroxyl,carboxyl and amino groups are realized by a multi-scale modeling method.The in fluences of the functional graphenes with di ff erent functional groups on the microstructure,mechanical and thermodynamic performances of polyimide-based composite models are investigated by the molecular dynamics simulation.The cell parameters,solubility parameters,elastic coefficients,Young’s moduli,shear moduli,and the values of glass-transition temperature(Tg)of polyimide-based composite models are calculated with the COMPASS force field.Moreover,the interaction energies and hydrogen bonds of composites are analyzed to explore the internal mechanisms for improving mechanical and thermodynamic properties.The results demonstrate that the density of PI matrix is 1.312 g.cm−3and the solubility parameter of PI matrix is 21.84 J1/2.cm−3/2,which are in accord with the actual PI parameters.The Young’s moduli of the composites increase obviously with the increase of the interaction energy between the PI matrix and the functional graphenes with hydroxyl,carboxyl and amino groups at 298 K and 1 atm.The Young’s moduli of PI and PI/graphene with carboxyl groups are respectively 3.174 GPa and 4.946 GPa and the shear moduli are respectively 1.139 GPa and 1.816 GPa.Comparing with pure PI/graphene composite,the average hydrogen bonds increase obviously after graphene has been functionalized.Because the interaction between the functional graphene and PI matrix increases,the movement of PI molecular chain needs more energy,and the rigidity of the composite is enhanced.TheTgof the composite also relates to the interaction energy.It is also found that theTgof the nano-composite e ff ectively decreases by the hybrid functional graphene.TheTgof pure PI is 663.57 K,while theTgvalues of PI/graphene and PI/graphene with carboxyl groups nanocomposites are 559.30 K and 601.61 K,respectively.Moreover,the density and interaction energy of hydrogen bonds of the PGCOOH are 784.81 kcal/mol and 1.396 g/cm3,respectively,which are the largest among their counterparts of the composite systems.The elastic coefficients show that the PGCOOH is more uniform than that other composites.All of these indicate that the graphene with carboxyl group can greatly enhance the interaction between graphene and PI,improve the mechanical properties and adjust theTgvalue of the PI matrix.The chemical modi fication of interaction energy in matrix is deemed to be of bene fit to the improvement in composite performance,and the interaction energy calculation is considered to be an e ff ective method of predicting the structures and performances of new composites.
polyimide,graphene,molecular dynamics,glass transition temperature
10.7498/aps.66.227101
∗国家自然基金(批准号:61372013)和黑龙江省自然科学基金(批准号:E201258)资助的课题.
†通信作者.E-mail:wlyang@hrbust.edu.cn
猜你喜欢
高中数理化(2022年14期)2022-08-15
纺织科技进展(2021年5期)2021-07-22
原子与分子物理学报(2019年5期)2019-04-28
植物营养与肥料学报(2019年12期)2019-03-07
橡胶工业(2016年2期)2016-02-23
中学化学(2015年12期)2016-01-19
中国塑料(2015年2期)2015-10-14
中国塑料(2015年8期)2015-10-14
中国塑料(2015年5期)2015-10-14
中国医学创新(2015年33期)2015-04-27
