
Na原子与惰性气体原子的相互作用势及光谱常数的理论计算
2017-12-08 02:11唐婷贵州大学明德学院贵州贵阳550002
化工管理 2017年34期
唐婷(贵州大学明德学院, 贵州 贵阳 550002)
Na原子与惰性气体原子的相互作用势及光谱常数的理论计算
唐婷(贵州大学明德学院, 贵州 贵阳 550002)
用量子化学CCSD(T)/aug-cc-pVTZ方法与基组,计算了van de Waals复合物NaNe和 NaAr复合物基态的相互作用势,并进行了基组重叠误差校正。采用Murrell-Sorbie函数进行非线性最小二乘法拟合,得到了NaX(X=Ne、Ar)复合物的势能函数解析表达式,确定了NaX(X=Ne、Ar)复合物的平衡结构和离解能,并进一步计算了这些复合物的光谱常数。
NaX(X=Ne,Ar);相互作用势;势能函数;光谱常数;基组重叠误差
1 引言
在许多物理过程中,相互作用势能具有及其重要的作用,一旦知道了势能,理论上所有可观测的物理量都可以通过解薛定谔方程求出[1]。许多研究者致力于碱金属原子与惰性气体原子间的相互作用势的研究,并从理论和实验中提出了多种可行的计算碱金属原子与惰性气体原子系统相互作用势能的方法[1-9]。
本文采用Gaussian 09软件包中的单双迭代(包含非迭代三重激发微扰)耦合簇CCSD(T)/aug-cc-pVTZ方法和基组对NaX(X=Ne、Ar)复合物在不同的核间距R进行逐点计算,研究其分子势能函数;然后用Murrell-Sorbie函数非线性最小二乘法拟合得到势能函数的具体形式,并计算出其光谱常数。为进一步研究Na原子和惰性气体原子相互作用体系的性质提供理论依据和数据参考。
2 计算方法
2.1 NaX(X=Ne、Ar)化合物间相互作用的计算

尽管在计算中使用的基函数是一个较大的基组,但仍然不是完备基集,基函数的重叠误差(BSSE)不可避免是存在的。从头算方法计算分子间的相互作用势能时,通常需要对基组误差进行校正。这里采用Boys和Bernardi的FCP(Full Couterpoise)方法来消除基函数重叠误差。在超分子近似中,FCP方法即要求用整个体系的基组在相应的几何构型下分别计算聚合体及各单体的能量,相互作用能则为相应能量的差值:
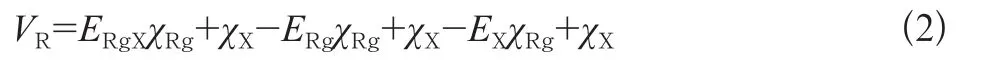
式中:χ表示基组。
将NaX复合物从头计算的未经基函数重叠误差校正的相互作用势能值V1(R)和经基函数重叠误差校正的相互作用势能值V2(R)随核间距R的变化如图1和图2所示。
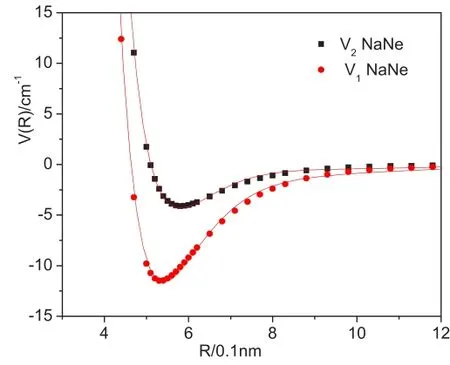
图1 NaNe体系相互作用的势能曲线

图2 NaAr体系相互作用的势能曲线
2.2 NaX(X=Ne、Ar)复合物光谱参数的计算
Murrell-Sorbie函数是常用于描述双原子分子的势能函数之一,其函数形式为:

首先用量子化学计算Gaussian09程序包中的单双迭代(包含非迭代三重激发微扰)耦合簇CCSD(T)/aug-cc-pVTZ方法和基组对NaX(X=Ne,Ar)复合物在不同的核间距R进行逐点计算,可得到一系列单点能量值。采用超分子近似,相互作用能为NaX(X=Ne、Ar)复合物的能量减去两个单体Na原子和惰性原子的能量,即:
式中:ρ=R-Re。Murrell-Sorbie函数中的参数具有明确的物理含义,De为离解能;Re为平衡核间距,并且参数a1、 a2和a3与相互作用的力常数密切相关。根据力常数f的定义:

即可得到二、三和四阶力常数f2、f3和f4与Murrell-Sorbie函数参数a1、 a2和a3的关系为:

力常数f2、 f3和f4与光谱常数ωe、 χeωe、Be、αe和D′e有如下关系:

用Murrell-Sorbie函数分别对未校正和经校正基函数重叠误差的相互作用势能值V1(R)和V2(R)进行非线性最小二乘法拟合,得到拟合相互作用势能函数的具体形式。将拟合势能函数参数代入(5)-(7)式计算二、三和四阶力常数f2、f3和f4及由(8)-(12)式,计算得到NaX(X代表He、Ne、Ar)化合物的光谱常数列于表1中。用Murrell-Sorbie函数拟合未校正和经校正基函数重叠误差的相互作用势能函数V1(R)和V2(R)的参数,未校正和经校正的NaX复合物的离解能De及平衡核间距Re都有一定的差别。分别计算出校正基函数重叠误差的离解能与未校正基函数重叠误差的解离能的相对误差Er(De)%和平衡核间距的相对偏差Er(Re)%列于表1中。
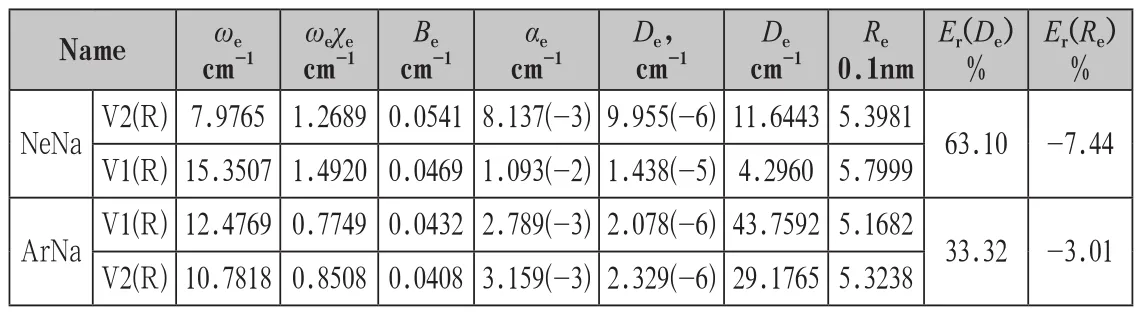
表1 NaX(X代表He、Ne、Ar)复合物的光谱参数及De和Re的相对误差Er%
3 结果讨论
从表1可以看出,用Murrell-Sorbie函数拟合未校正和经校正基函数重叠误差的相互作用势能函数 和 的参数,NeNa复合物未校正和经校正的离解能De分别为11.6443cm-1和4.2960cm-1。经校正基函数重叠误差的离解能是未校正基函数重叠误差的63.10%,说明基函数重叠误差对计算离解能的影响是不容忽视的。NeNa复合物势能函数 和 的平衡核间距Re分别为0.5398nm和0.5799nm,两平衡核间距并不相同,其未校正基函数重叠误差的平衡核间距与校正基函数重叠误差的间距的相对偏差为-7.44%。
ArNa复合物未校正和经校正的离解能De分别为29.17cm-1和11.64cm-1。经校正基函数重叠误差的离解能是未校正基函数重叠误差的33.32%,说明基函数重叠误差对计算离解能的影响是不容忽视的。ArNa复合物势能函数 和 的平衡核间距Re分别为0.5168nm和0.5323nm,两平衡核间距并不相同,其未校正基函数重叠误差的平衡核间距与校正基函数重叠误差的间距的相对偏差为-3.01%。
在表1中,本文用两个势能函数计算了NaX(X=Ne、Ar)复合物的光谱常数,希望能与实验值进行比较,同时对研究NaX(X=Ne、Ar)复合物的其它性质提供参考信息。
[1]王君.锂与惰性气体相互作用势计算方法及组合规则的研究[D].成都:四川大学,2007.
[2]Patil S H.Adiabatic potentials for alkali-inert gas systems in the ground state [J].J.Chem.Phys.,1991,94: 8089
[3]Kleinekath-fer U,Tang K T,Toennies J P.Yiu C L.Potentials for some rare gas and alkali-helium systems calculated from the surface integral method [J].Chem.Phys.Lett.,1996,249: 257-263.
[4]Kleinekat hofer U,Tang K T,Toennies J P.et al.Potentials for some rare gas and alkali2helium systems calculated from the surface integral met hod[J].Chem.Phys.Lett.,1996.249:257~263.
[5]杨向东,牟致栋,等.Li-He与Li-Xe相互作用势的研究[J].四川大学学报(自然科学版),1997,34(3):292.
[6]王君,杨缤维,等.锂与惰性气体相互作用势组合规则的研究[J].四川大学学报(自然科学版),2006,43(4):832-835.
[7]李萍,姜明.锂原子与部分惰性气体原子间相互作用势的计算[J].原子与分子物理学报,2000,17(2):306-307.
[8]李劲,令狐荣锋,等.Li、Na、K与He原子系统相互作用势的理论研究[J].四川大学学报(自然科学版),2008,45(5):60-65.
[9]李萍,姜明.钠原子与氖、氩、氪和氙原子间相互作用势的面积分方法计算[J].原子与分子物理学报,1998.增刊.
猜你喜欢
——《势能》
文化纵横(2022年3期)2022-09-07
中学生数理化·八年级物理人教版(2022年6期)2022-06-05
中学生数理化·八年级物理人教版(2021年6期)2021-11-22
中学生数理化·八年级物理人教版(2019年6期)2019-06-25
中成药(2018年7期)2018-08-04
中成药(2018年3期)2018-05-07
科教导刊(2017年23期)2017-10-10
中华建设科技(2017年6期)2017-07-21
中成药(2017年5期)2017-06-13
中国医药科学(2014年17期)2014-09-26
