
伴强直发作的Dravet综合征2例报告并文献复习
2018-03-30 01:43魏春苗夏桂枝任榕娜
东南国防医药 2018年2期
魏春苗,夏桂枝,任榕娜
0 引 言
Dravet综合征(Dravet syndrome,DS)是一种少见的婴儿期起病的难治性早发性癫痫脑病。发病率为1/22 000~1/40 000[1-2],死亡率高且有癫痫猝死的风险[3-4]。一方面DS患儿发病前生长发育正常,1岁以内以热性惊厥起病,早期神经影像学及脑电图无异常发现,诊断困难;另一方面DS患儿对抗癫痫药物疗效差,1岁以后多出现精神运动发育落后或倒退且症状可持续到成年;因此早期诊断、合理治疗尤为重要。近年来SCN1A基因突变的发现为DS的早期诊断提供了重要依据,对合理用药也具有一定的指导作用。现将2例伴强直发作的DS少见表型患儿的临床及基因突变特点进行分析,结合文献复习报道如下。
1 资料与方法
1.1一般资料收集2011年1月至2017年3月在福州总医院儿科神经专科门诊及病房诊治的2例伴强直发作的DS患儿的临床资料,包括发病年龄、性别、临床表现、个人史、家族史、实验室检查、视频脑电图,头颅MRI、诊疗过程及随访情况。
病例1:7岁女性患儿,足月顺产,父母之间无血缘关系,无癫痫及热性惊厥家族史。5月龄时首次于体温38 ℃左右出现惊厥发作,表现为意识丧失,四肢阵挛性抽动,持续约10~15 min自行缓解。此后反复出现发热或感染后类似发作,2~3岁发作频繁,3岁以后发作次数减少,约3个月发作1次。 4岁时出现3次不典型失神发作。5岁后出现反复强直发作,表现为意识丧失,双眼上翻,四肢强直,头后仰,持续5~8 min自行缓解,缓解后全身无力,意识逐渐恢复,发作前有热或无热,每年2~3次。曾先后给予托吡酯、左乙拉西坦、丙戊酸钠、奥卡西平、氯硝西泮等药物治疗,发作控制不佳,现用丙戊酸钠联合托吡酯治疗。首次发作及发作1年后2次查头颅MRI及视频脑电图(vedio electroencephalogram,VEEG)未见异常。6岁时VEEG:①基本波率为5~7次/s慢波节律,波形不整,左右波幅不对称,调节调幅差,两颞枕区慢波节律优势,时而高波幅同步阵发;②异常脑波:中-高波幅同步阵发慢波节律,两侧波幅不对称,两额颞、(旁)中线区夹杂尖棘慢波。血电解质、血氨、血浆乳酸、血尿遗传代谢病筛查均未见异常。8个月时Gesell发育诊断量表评估:99;6岁韦氏儿童智能测试:85,感觉统合严重失调;现上小学,学习成绩较差。
病例2:3岁5个月男性患儿,足月顺产,父母之间无血缘关系,无癫痫及热性惊厥家族史。首次于8月龄感冒后发热,体温38.5 ℃左右出现右侧肢体阵挛性抽动,随后出现意识丧失、双眼上翻、牙关紧闭、口唇青紫、四肢强直阵挛,前后持续约15 min自行缓解,次日再次发作1次,以后间隔几天~1个月发作1次,有时1天数次,1~3岁发作次数稍有减少,间隔2~3个月1次,有热或无热,表现同前。3岁后出现2次反复强直发作,表现为意识丧失、双眼上翻、牙关紧闭、四肢强直呈角弓反张状,无阵挛出现,约5 min后自行缓解,缓解后全身无力,意识逐渐恢复。10个月查头颅MRI、VEEG未见异常;2岁时VEEG:①两侧波幅不对称、调节调幅差,同步阵发4~6 Hz慢波节律,两后头部(旁)中线区优势;②广泛的尖棘慢波节律,旁中线区优势。2个月前复查VEEG无明显变化。血电解质、血氨、血浆乳酸、血尿遗传代谢病筛查均未见异常。曾先后口服左乙拉西坦、丙戊酸及托吡酯治疗,疗效不佳,现用丙戊酸联合托吡酯治疗。10个月Gesell发育诊断量表评估:95;2岁Gesell发育诊断量表评估:80;现走路易跌倒;语言发育明显落后。
1.2方法采集患儿及父母外周血2 mL(EDTA抗凝),用BloodGen Midi Kit (CWBIO, China) 提取患儿全基因组DNA,操作按照试剂盒说明书进行。参考文献与OMIM数据库信息,将OMIM数据库中所有与四千种单基因遗传病相关的基因组外显子区域定制罗氏NimbleGen捕获探针,然后制备捕获文库、Illumina平台进行二代测序,最后进行数据分析。根据所验证位点序列设计引物,采用PCR方法进行扩增,ABI 3730XL 测序仪进行一代测序验证,测序引物采用原PCR引物,然后进行基因序列分析和比对。二代测序和一代测序验证均委托北京智因东方转化医学研究中心完成。
2 结 果
2.1临床特点2例患儿分别在出生5个月和8个月起病,热性惊厥为其首次发作表现。发作形式中除有常见的全身及偏侧阵挛、强直阵挛和不典型失神等多种发作外,都出现了DS少见的强直发作。2例患儿均无癫痫及热性惊厥家族史,实验室检查及头颅MRI未见异常,视频脑电图为DS特征性棘慢波发放。2例患儿先后应用多种抗癫痫药物,但疗效均不佳,起病前发育正常,现都有不同程度的精神运动发育落后。
2.2基因突变特点病例1:四千种单基因病基因检测结果:SCN1A变异,chr2:166904182,c.1125G>C(E10),P.315.L>F,杂合,其父母均未发现相同突变,为新发错义突变,见图1a。病例2:四千种单基因病基因检测结果:SCN1A变异,chr2: 166894356,c.2876G>C(E17),P.959,C>S,杂合,父母均未发现相同突变,为新发错义突变,见图1b。
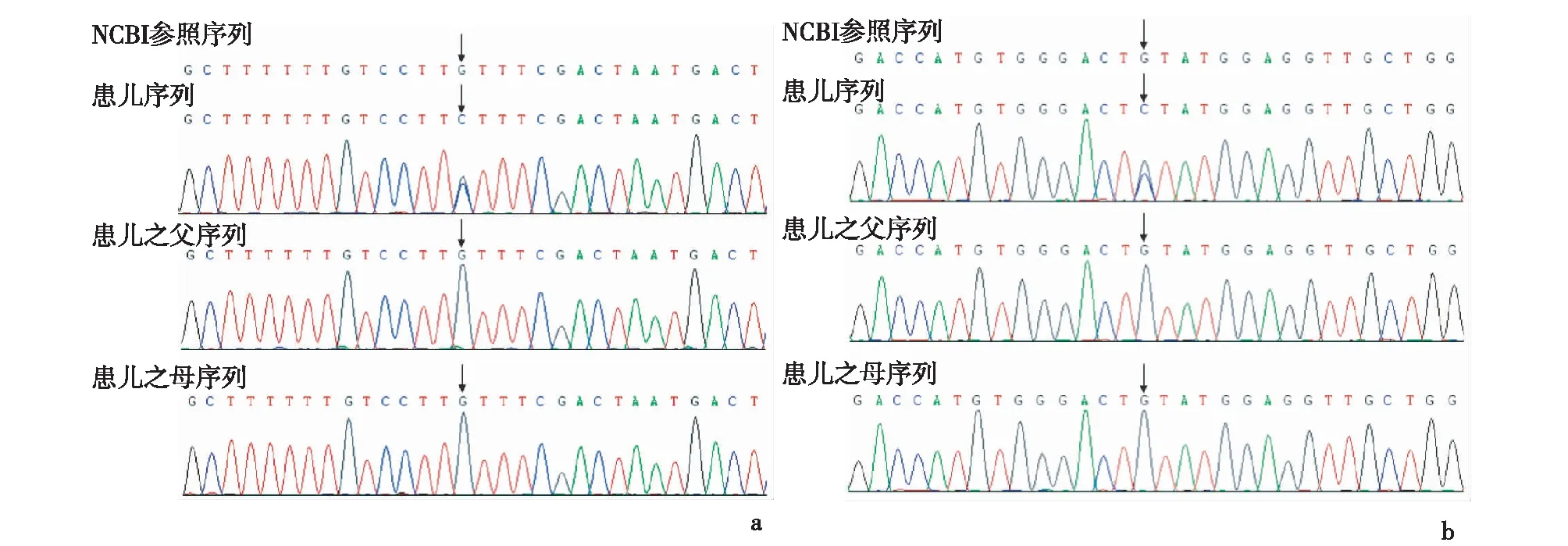
a:病例1(c.1125G>C);b:病例2(c.2876G>C)图1 Dravet综合征SCN1A突变一代测序峰图(箭头所指为突变位点)
3 讨 论
DS的临床核心表现为:①1岁以内起病,首次发作为持续时间较长的热性惊厥,或疫苗接种、洗热水澡等诱发的无热惊厥;②1岁后逐渐出现多种形式的无热惊厥,包括全身或半侧阵挛、强直阵挛发作、肌阵挛发作、不典型失神、局部性发作等,极少数病例出现强直发作,常发展为癫痫持续状态;③首次发作时生长发育正常,1岁后逐渐出现精神运动发育落后或倒退,可出现共济失调、锥体束征及运动不协调;④神经影像学多无异常;⑤脑电图检查在1岁以前多无异常,以后逐渐出现θ波为主的慢活动或广泛性、局灶性的癫痫放电;⑥对抗癫痫药物治疗效果差[5-6]。DS的诊断主要依据上述核心症状。
特别的是本报道中2例患儿在病程中均出现了强直发作这一DS少见的发作形式,表现为轴性强直,例1在5岁后出现,例2在3岁后出现,均在癫痫频繁发作期后,发作次数较前明显减少。在对DS的首次描述中并未提及强直发作[7],之后表现为强直发作的DS病例陆续被报道,但例数均较少[8-9],最新的文献对DS核心症状的描述中,仍然认为强直发作是不常见的发作形式[10-11]。强直发作表现类似Lennox-Gastaut综合征(LGS)的轴性强直,有时伴有肌阵挛发作,强直发作往往是散发的,在一次发作过程中反复重复出现。Dravet等[8]报道的发作期脑电图表现为电活动减少或低波幅的快波节律后出现慢波或间隔以电活动减少的快速的募集节律,发作间期睡眠脑电图表现为与LGS相同的快波节律和多棘波发放。Nabbout等[9]报道强直发作间期脑电图为前额区的慢的双向或三向棘波,后伴或不伴有慢波。本报道中2例患儿未记录到上述强直发作的脑电图表现,但临床表现与报道的DS的强直发作基本相同。
目前对DS研究最大的进展是DS基因突变的发现。2001年Claes等[12]检测出7例DS患儿存在编码钠离子通道α亚单位的SCN1A基因新发突变,随后的研究发现70%~80%的DS患儿携带SCN1A基因突变[9,13],这些突变发生于SCN1A不同的基因位点,90%为新发突变,有截断突变、错义突变、无义突变、缺失突变等多种突变形式[14]。虽然到目前为止,基因型和表现型之间的关系尚未阐明,并不是所有携带SCN1A基因突变的都是DS,但SCN1A基因突变对于DS尤其是少见表型病例的早期诊断仍是一个非常有力的诊断依据[9]。本报道中2例患儿出现DS少见的强直发作,早期诊断更加困难,最终基因检测出SCN1A基因新发错义突变而确诊,可见基因检测对于DS的早期诊断至关重要。近年来在一些临床表现类似DS而SCN1A基因突变阴性的女性患儿中检测出位于X染色体上的编码钙粘蛋白的PCDH19基因突变[15]。因此,对于女性患儿如SCN1A基因突变检测阴性,应进行PCDH19基因突变检测。其他如SCN1B、GABRA1和STXBP1等基因突变导致的DS,亦有报道[16-17]。
对所有抗癫痫药物抵抗是DS的一个重要特征,目前对DS的治疗目标是防止长时间的惊厥发作,减少惊厥发作次数,改善患儿的精神运动发育。因大多数DS患儿携带编码钠离子通道α亚单位的SCN1A基因突变,钠离子通道阻滞剂如卡马西平、奥卡西平、拉莫三嗪、苯妥因可使病情加重,苯妥因还可诱发手足徐动症,应避免使用。氨己烯酸也可使病情恶化,苯巴比妥和卢非酰胺作用甚微。在疑似DS的首次无热或复杂性热性惊厥发作后给予丙戊酸治疗目前已经达成共识。DS的病理生理机制被普遍接受的是中间神经元假说,即SCN1A基因突变使抑制性的GABA能神经元被抑制而导致兴奋性神经元的过度兴奋[18],司替戊醇具有增强GABA能神经元的作用,可以缩短发作时间并预防SE的发生[19],很多文献报道托吡酯对部分患儿可以减少发作,因此反复惊厥发作可加用托吡酯或司替戊醇,如丙戊酸加托吡酯或司替戊醇效果不佳,可以将托吡酯和司替戊醇互相替换,或替换成溴化物。因为司替戊醇是细胞色素P450的抑制剂,加用司替戊醇时要适当减少丙戊酸的用量。在药物治疗无效的情况下可以选用生酮饮食治疗[20]。本报道中2例患儿应用过多种抗癫痫药物均不能控制发作,在确诊DS前例1患儿在托吡酯、左乙拉西坦、丙戊酸钠治疗无效的情况下,曾短时间应用过钠离子通道阻滞剂奥卡西平。文献报道约1/3的DS患儿曾有钠离子通道阻滞剂的应用经历[21],早期进行SCN1A基因突变检测不仅可以帮助DS患儿早期明确诊断,还可以避免钠离子通道阻滞剂等可能加重病情的药物的应用。2例患儿目前均口服丙戊酸联合托吡酯治疗,癫痫虽未完全控制,但发作次数较前减少,持续时间也较前缩短,均未出现癫痫持续状态。
通过回顾性分析上述2例患儿的临床和基因突变特点,可以发现DS患儿在病程中的癫痫频繁发作期后可以出现强直发作形式;除此以外,2例患儿在病史、临床表现、对抗癫痫药物的反应以及预后方面与其他无强直发作的DS患儿基本相同。基因检测有助于这种少见表型DS的早期确诊及选用合适的抗癫痫药物治疗。
[1] Wu YW, Sullivan J, McDaniel SS,etal. Incidence of Dravet syndrome in a US population[J]. Pediatrics, 2015, 136(5): 1310-1315.
[2] Bayat A, Hjalgrim H, Moller RS,etal. The incidence of SCN1A-related Dravet syndrome in Denmark is 1:22000: a population-based study from 2004 to 2009[J]. Epilepsia, 2015, 56(4): 36-39.
[3] Shmuely S, Sisodiya SM, Gunning WB,etal. Mortality in Dravet syndrome: a review[J]. Epilepsy Behav, 2016, 64(Pt A): 69-74.
[4] 黄逸青,吴 原.癫痫猝死模型的研究进展[J].医学研究生学报,2016,29(1):100-103.
[5] Dravet C. Dravet syndrome history[J]. Dev Med Child Neurol, 2011, 53( Suppl 2):1-6.
[6] Scheffer IE. Diagnosis and long-term course of Dravet syndrome[J]. Eur J Paediatr Neurol, 2012, 16( Suppl 1): S5- S8
[7] Dravet C. Les e’pilepsies graves de l’enfant[J]. Vie Med,1978, 8:543-548.
[8] Dravet C. The core Dravet syndrome phenotype[J]. Epilepsia, 2011, 52(Suppl 2):3-9.
[9] Nabbout R, Desguerre I, Sabbagh S,etal. An unexpected EEG course in Dravet syndrome[J]. Epilepsy Res, 2008, 81(1):90-95.
[10] Connolly MB. Dravet syndrome: Diagnosis and long-term course[J]. Can J Neurol Sci, 2016, 43 (Suppl 3): S3-S8.
[11] Gataullina S, Dulac O. From genotype to phenotype in Dravet disease[J]. Seizure, 2017,44:58-64.
[12] Claes L, Del-favero J, ceulemans B,etal. De nove mutation in the sodium channel gene SCN1A cause severe myoclonic epileptic in fancy[J]. Am J Hum genet, 2001, 68(6):1327-1332.
[13] 许小菁, 张月华, 孙慧慧, 等. Dravet综合征SCN1A基因突变的遗传特点及表型研究[J].中华医学遗传学杂志, 2012, 29(6): 625-630.
[14] Depienne C, Trouillard O, Saint-Martin C,etal. Spectrum of SCN1A gene mutations associated with Dravet syndrome: analysis of 333 patients[J]. Med Genet, 2009, 46(3): 183-191.
[15] Depienne C, Bouteiller D, Keren B,etal. Sporadic infantile epileptic encephalopathy caused by mutations in PCDH19 resembles Dravet syndrome but mainly affects females[J]. PLoS Genet, 2009, 5(2):e1000381.
[16] Patino GA, Claes LR, Lopez-Santiago LF,etal. functional null mutation of SCN1B in a patient with Dravet syndrome[J]. J Neurosci, 2009, 29(34):10764-10778.
[17] Carvill GL, Weckhuysen S, McMahon JM,etal. GABRA1 and STXBP1: novel genetic causes of Dravet syndrome[J]. Neurology, 2014, 82(14):1245-1253.
[18] Cheah CS, Yu FH, Westenbroek RE,etal. Specific deletion of NaV1.1 sodium channels ininhibitory interneurons causes seizures and premature death in amouse model of Dravet syndrome[J]. Proc Natl Acad Sci USA, 2012, 109(36):14646-14651.
[19] Grosenbaugh DK, Mott DD. Stiripentol is anticonvulsant by potentiating GABAergic transmission in a model of benzodiazepine-refractory status epilepticus[J]. Neuropharmacology, 2013, 67:136-143.
[20] Wirrell EC. Treatment of Dravet syndrome[J]. Can J Neurol Sci,2016, 43 (Suppl 3): S13-S18.
[21] Aras LM, Isla J, Mingorance-Le Meur A. The European patient with Dravet syndrome: results from a parent-reported survey on antiepileptic drug use in the European population with Dravet syndrome[J].Epilepsy Behav, 2015, 44:104-109.
猜你喜欢
中国民间疗法(2021年10期)2021-07-22
中国医院用药评价与分析(2021年2期)2021-04-23
医药前沿(2019年11期)2019-01-05
中国卫生标准管理(2015年18期)2016-01-20
中国卫生标准管理(2015年5期)2016-01-14
中国学术期刊文摘(2015年8期)2015-10-29
现代医院(2015年11期)2015-02-21
卫生职业教育(2014年14期)2014-05-16
四川生理科学杂志(2014年2期)2014-02-28
海南医学(2012年17期)2012-04-08
