
深海真菌Dichotomomyces cejpii胶霉毒素生物合成基因启动子的克隆和功能鉴定
2018-05-07 08:33黄自磊章卫民叶伟李赛妮李浩华朱牧孜
生物技术通报 2018年4期
黄自磊 章卫民 叶伟 李赛妮 李浩华 朱牧孜
(1. 中国科学院南海海洋研究所,广州 510301;2. 广东省微生物研究所 省部共建华南应用微生物国家重点实验室 广东省菌种保藏与应用重点实验室 广东省微生物应用新技术公共实验室,广州 510070;3. 中国科学院大学,北京 100049)
胶霉毒素(Gliotoxin)主要是由烟曲霉(Aspergillus fumigatus)产生的一类含硫二酮哌嗪(ETPs)类化合物,能够结合含硫蛋白,破坏细胞内的氧化还原反应平衡[1-3]。胶霉毒素具有广谱抑菌、抑病毒、抗肿瘤和免疫调节活性,也可制备成杀菌剂和除草剂防治植物病害,有很好的应用前景[4]。据报道,GliG、GliI和GliO是烟曲霉胶霉毒素生物合成所必须的功能基因[5]。启动子作为结构基因和功能基因转录调控的必要元件,能够募集转录因子与RNA聚合酶精确的起始转录[6-7]。近年来,丝状真菌在新型、高活性次级代谢产物的发掘以及活性酶的研究和开发方面发展迅速,且丝状真菌中启动子对其内源基因的转录活性较高,因此很多不同种属丝状真菌的启动子被发掘出来,如木霉属(Trichoderma)pki1启动子,里氏木霉(T. reesei)的cbh1、tef1启动子和gpdA启动子,曲霉属(Aspergillus)的glaA启动子等[8-9]。本课题组前期从深海真菌Dichotomomyces cejpii中分离得到多个胶霉毒素,它们具有良好的生物活性[10],进一步对该真菌进行了转录组测序,结果表明基因GliG、GliI和GliO分别编码谷胱甘肽硫转移酶、转氨酶和醛还原酶。目前,关于深海真菌D. cejpii中胶霉毒素的生物合成基因及其启动子的功能未见报道。本研究采用染色体步移技术对D. cejpii中胶霉毒素的生物合成关键基因GliG、GliI、GliO启动子进行克隆,并采用荧光素酶报告基因载体和潮霉素抗性基因表达载体进行功能验证,从而为后期通过转录调控和异源表达以获得更多高活性的新型胶霉毒素奠定分子生物学基础。
1 材料与方法
1.1 材料
1.1.1 试剂和仪器 DNA胶回收试剂盒、质粒小提试剂盒(天根生化试剂公司,北京),HiPure Plant RNA Mini Kit(美基生物科技有限公司,广州),Genome walking kit、PrimeSTAR MAX Premix(TaKaRa,大连),2x Taq master mix(Microanalysis生物科技公司,北京),DNA Marker 2k、trans 2k plus II、1k(全式金生物技术有限公司,北京),All-in-one RT Master Kit(Abm, 加 拿 大 ),TransStart Tip Green qPCR SuperMix(+Dye II)(全式金生物技术有限公司,北京),细胞超声破碎仪(Sonics,美国),GloMax 20/20荧光光度计(Promega,美国),荧光素酶报告基因检测试剂盒(碧云天生物技术有限公司,上海)
1.1.2 菌株和载体 深海真菌Dichotomomyces cejpii分离自南海沉积物[11],pGL3-Basic载体(淼灵生物科技有限公司,武汉),pAN7-1质粒(淼灵生物科技有限公司,武汉),Trans5α感受态细胞(全式金生物技术有限公司,北京),BL21(DE3)感受态细胞(全式金生物技术有限公司,北京),pEASY-T1 Cloning Kit(全式金生物技术有限公司,北京),酿酒酵母BY4742,保藏于广东省微生物研究所。
1.2 方法
1.2.1D. cejpii转录组测序 将深海真菌D. cejpii接种于YPD培养基平板,37℃培养72 h,挑取新鲜的菌丝体,利用植物RNA提取试剂盒提取RNA,利用All-in-one RT Master Kit逆转录获得cDNA,并采用Hiseq2000进行转录组测序,通过前期转录组测序得到的目的基因序列设计基因GliG、GliI、GliO引物,引物序列为:GliG(FP:5'-atgaccgaacgacc ttcttgatctcg-3',RP :5'-caatagtccatactccttctcgcc-3'),GliI(FP :5'-atgcctcacgcagaaacactcccc-3',RP :5'-ccacctcttatccacccccaatg-3'),GliO(FP:5'-atggccaattctcgacccaacatcg-3',RP:5'-gttgaagttatccaggattgcg-3'),以cDNA为模板进行荧光定量PCR,分析这3个基因的相对表达量,并通过琼脂糖凝胶电泳进一步验证目的基因的表达水平。
1.2.2GliG、GliI、GliO启动子的克隆和序列分析 根据前期转录组测序结果,利用Genome walking kit试剂盒,设计GliG、GliI、GliO序列中特异性反向引物(表1),并利用正向引物AP3进行巢式PCR扩增,并将最后一步的PCR片段利用pEASY-T1试剂盒进行TA克隆,转化至trans5α感受态细胞,涂布于氨苄青霉素抗性平板筛选出阳性克隆,利用T7-F和T7-ter引物进行菌液PCR验证阳性克隆并测序,获得目的基因GliG、GliI、GliO上游启动子序列,并利用启动子预测软件(http://www.fruitfly.org/seq_tools/promoter.html)分析启动子序列,获得启动子的核心区域。
1.2.3 启动子-pGL3-Basic载体及启动子-pAN7-1-2μ复制子载体构建 根据克隆得到的GliG上游核心启动子片段以及pgpd启动子,设计单一酶切位点的引物,pgpd启动子上下游引物分别加酶切位点XhoI和Hind III,GliG核心启动子上下游引物分别加酶切位点NheI和Hind III,与pGL3-Basic载体对应的酶切产物连接转化构建载体。GliI/GliO核心启动子通过环形聚合酶延伸克隆(Circular polymerase extension coloning,CPEC)技术设计同源臂引物,与线性载体进行重叠延伸PCR构建载体[12]:设计GliI/GliO启动子核心区域引物以及pGL3-Basic载体引物,保证载体和片段有20 bp重叠区域,利用Max酶进行三步PCR反应,程序为:98℃ 10 s,55℃ 15 s,72℃ 1 min,克隆出目的片段和载体片段,胶回收试剂盒纯化,并将克隆出的启动子核心目的片段以及pGL3-Basic载体片段各1 μL(浓度为50 ng/μL以上)用于10 μL PCR体系中,不加引物,进行环化连接克隆,转化trans5α,利用菌落PCR筛选阳性克隆并测序,验证载体构建结果。GliG上游核心启动子区域和2μ-pAN7-1载体的构建:先利用CPEC法将酵母2μ复制子插入到pAN7-1载体中,然后利用酶切连接的方法,对GliG核心启动子设计BglII和PshA I酶切位点的上下游引物,PCR扩增并酶切,与酶切后的2μ-pAN7-1载体连接转化,将目的基因GliG上游核心启动子区域取代pgpdA启动子,构建2μ-pAN7-1-GliG核心启动子载体,并电转入酿酒酵母细胞中,利用不同浓度(0、50、100、150、200 μg/mL)潮霉素抗性平板进行筛选和验证。

表1 目的基因GliG、GliI、GliO特异性反向引物序列
1.2.4 启动子荧光素酶活性检测 将构建成功的含有目的基因启动子的重组pGL3-Basic质粒载体,空载体pGL3-Basic(阴性对照)和pgpd-pGL3-basic载体(阳性对照,pgpd是真菌双孢蘑菇中分离鉴定的启动子[13],能有效启动目的基因的表达)分别转化到trans5α感受态细胞,利用氨苄青霉素抗性平板筛选阳性克隆,并测序验证。将阳性克隆扩大培养,利用质粒小量提取试剂盒提质粒,并转化BL21(DE3)感受态细胞,挑取单菌落并保种。含有pGL3-basic-空载/GliG/GliI/GliO/pgpd质粒的trans5α以及BL21(DE3)感受态细胞分别于20 mL LB液体培养基中扩大培养,37℃、200 r/min摇床培养,当OD值为0.8-1.0,8 000 r/min离心5 min取沉淀,并用PBS缓冲液洗涤2-3次,最后用2 mL的PBS缓冲液溶解,并进行细胞超声破碎(振幅 40%,工作5 s/停顿5 s)10 min,10 000 r/min离心取上清置于冰上。取上清100 μL和萤火虫荧光素酶检测试剂100 μL与1.5 mL EP管中混匀,用荧光光度计检测不同重组菌上清的RU值,反应时间为5 s。
1.2.5 潮霉素抗性基因表达验证 用不同浓度(0、50、100、150、200、250 μg/mL)的潮霉素抗性平板对酿酒酵母抗性进行筛选,30℃培养3 d,确定潮霉素对于酿酒酵母的最适筛选浓度。制备酿酒酵母感受态细胞[14],将 2μ-pAN7-1 载体以及 2μ-pAN7-1-GliG载体分别电转入酿酒酵母细胞中(1 500 V,5 ms),均匀涂布于潮霉素抗性平板中,并利用菌落PCR筛选阳性克隆,并进一步测序验证。
2 结果
2.1 GliG、GliI、GliO的表达水平分析
以D. cejpii的cDNA为模板进行荧光定量PCR,结果表明,目的基因GliG的相对表达水平显著高于GliI基因和GliO基因(P<0.01)(图1-A)。琼脂糖凝胶电泳结果表明,GliG对应条带明显亮于GliI、GliO对应条带(图1-B),说明GliG基因表达水平相对较高,在D. cejpii胶霉毒素生物合成过程中发挥重要作用。
2.2 GliG、GliI、GliO启动子的克隆分析
通过3-4次巢式PCR反应对D. cejpii基因组扩增,得到对应目的片段,I4(GliI启动子第4轮扩增)约2.5 kb,O4(GliO启动子第4轮扩增)约为3.1 kb,G3(GliG启动子第3轮扩增)约为4 kb(图2)。菌液PCR筛选得到阳性克隆G1、I1、I2、O1、O2(图3),通过与目的基因序列比对,证实均为包含目的基因启动子片段。

图1 GliG、GliI、GliO的表达水平
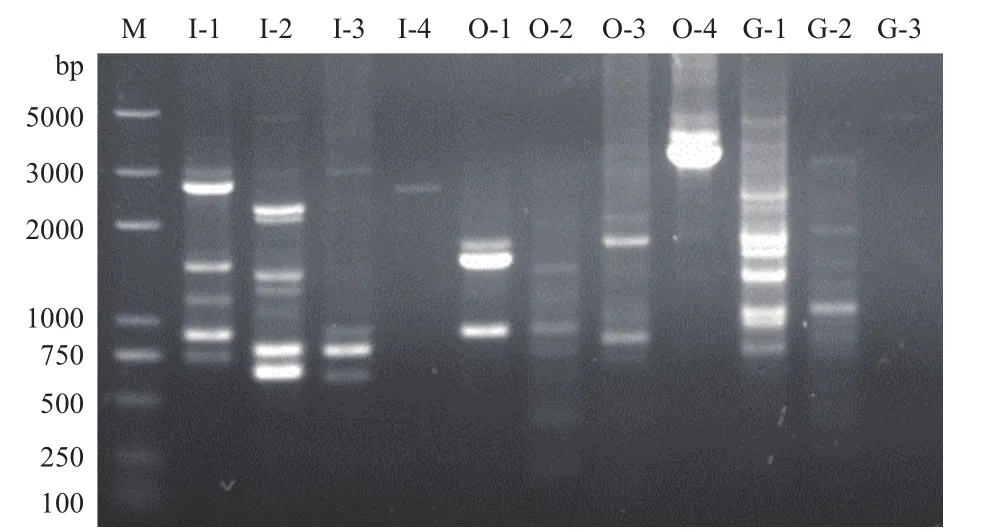
图2 GliI、GliO、GliG染色体步移扩增得到的目的片段
将GliG、GliI、GliO三个基因的上游2-3 kb序列在启动子预测网站(http://www.fruitfly.org/seq_tools/promoter.htmL)寻找启动子核心区域(表2),分值相对较高(最高为1),然后扩增出这3个基因的上游区域,结果GliG基因上游500 bp,GliI基因上游700 bp,GliO基因上游300 bp(图4)。
2.3 pGL3-basic-启动子载体构建以及荧光素酶活性检测
利用CPEC方法成功构建了4个分别含有GliG、GliI、GliO、pgpd核心启动子的pGL3-Basic重组表达载体,并利用氨苄青霉素平板筛选,挑取单菌落进行菌落PCR筛选阳性克隆,测序验证成功(图5)。

表2 目的基因GliG、GliI、GliO启动子核心序列

图4 GliG、GliI、GliO、pgpd核心启动子
将含有不同启动子核心序列的pGL3-Basic重组载体转化至大肠杆菌,对重组菌中的荧光素酶进行RU检测。结果(图6)表明,含有GliG启动子的重组菌中荧光素酶活性远高于其他重组菌,说明GliG核心启动子区域具有较高的转录活性。

图5 含pGL3-basic-GliG、GliI、GliO核心启动子的菌落PCR验证

图6 pGL3-basic-空载/pgpd/GliG/GliI/GliO核心启动子载体在Trans5α和BL21细胞中的荧光素酶RU值
2.4 GliG-pAN7-1质粒的构建及潮霉素抗性的验证
用GliG核心启动子区域替换pgpdA启动子,构建得到重组载体,并测序成功(图7)。将酿酒酵母均匀涂布至不同浓度的潮霉素抗性平板,发现在100 μg/mL浓度以上的潮霉素抗性平板上酿酒酵母的生长受到明显抑制(图8),在200 μg/mL的潮霉素平板上基本上不生长,从而确定最适宜的筛选浓度为 200 μg/mL。
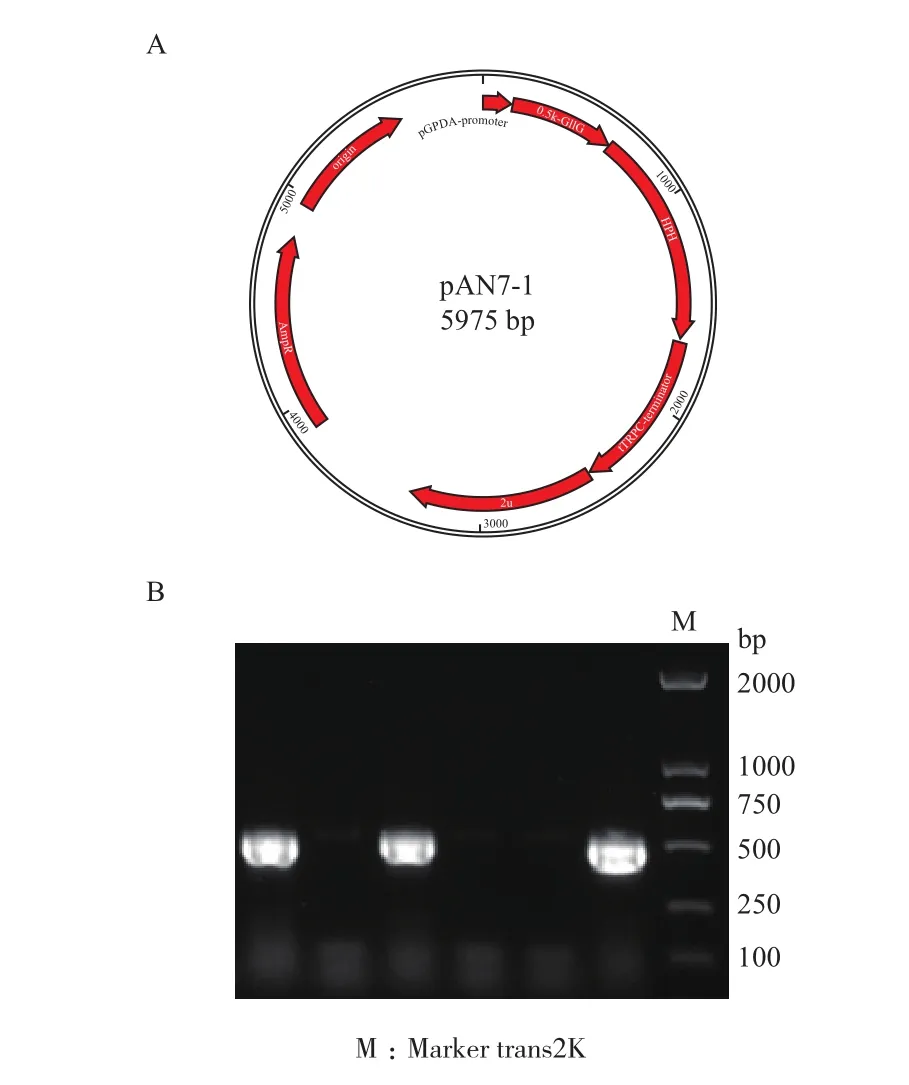
图7 2μ-pAN7-1-GliG核心启动子载体以及trans5α菌落PCR验证

图8 不同浓度的潮霉素抗性平板筛选酿酒酵母
将2μ-pAN7-1-GliG核心启动子载体以及2μ-pAN7-1载体(阳性对照)分别电转入酿酒酵母,与阴性对照和阳性对照相比,发现在200 μg/mL的潮霉素平板上,含有2μ-pAN7-1-GliG核心启动子载体的酵母生长较快,而且菌落数较多(图9)。挑取含有重组载体的酿酒酵母进行菌落PCR,筛选得到阳性克隆,并通过测序得到验证(图10)。
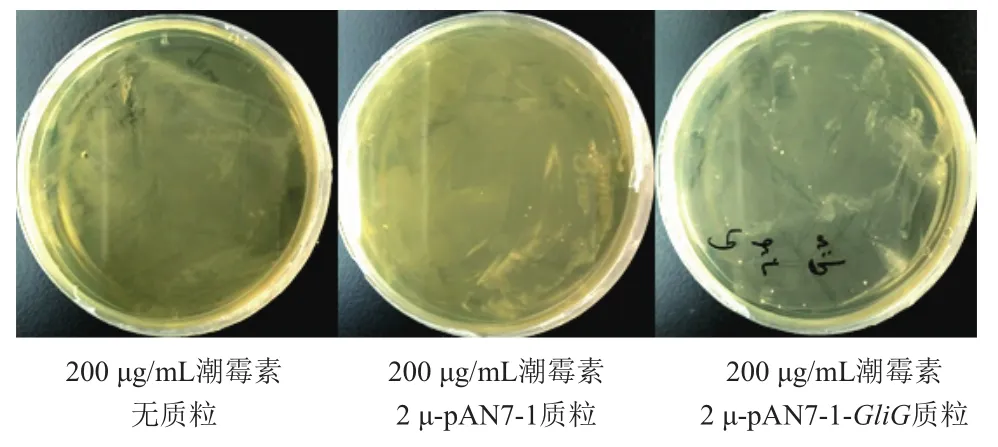
图9 2μ-pAN7-1-GliG核心启动子电转酿酒酵母筛选(培养3 d)

图10 2μ-pAN7-1-GliG核心启动子酿酒酵母菌落PCR验证
3 讨论
真菌中次级代谢产物的生物合成关键酶需要活性较强的启动子来转录调控,尤其是对于一些关键限速酶,启动子的活性对于关键酶的高水平表达和代谢产物的高效生物合成至关重要[13]。在丝状真菌中许多功能性的启动子已经应用于内源和外源蛋白的表达,例如烟曲霉属真菌中启动子用于异源表达人类白介素hIL-6,人血清白蛋白,人层粘连蛋白等,且产量大幅度提高[15-16]。Dolan等[5]首次对烟曲霉中胶霉毒素的生物合成基因簇及其代谢通路进行了详细的阐述,其中GliG、GliI、GliO三个基因是胶霉毒素合成家族gli簇中不可缺少的关键基因,在胶霉毒素合成过程中,GliG基因编码的谷胱甘肽S转移酶是不可缺少的关键酶。目前,深海真菌D. cejpii中胶霉毒素生物合成基因的启动子及其功能未见报道。本研究通过对该真菌中gli簇关键基因启动子进行了克隆和功能预测,发现GliG、GliI、GliO启动子对这三个基因具有转录活性,其中GliG启动子的转录活性最强,提示该启动子在胶霉毒素生物合成中发挥重要作用。本研究结果为分析gli簇基因家族基因的转录调控机制,寻找关键转录调控因子,并为后续激活D. cejpii中隐性或低活性的关键基因或基因簇,生物合成新型的胶霉毒素衍生物奠定基础。同时,本研究通过对GliG基因启动子的研究,还找到了起始转录GliG的启动子序列,并能在酵母细胞中发挥作用,对潮霉素抗性基因能进行有效地起始转录和表达,提示该启动子可用于表达异源或外源谷胱甘肽硫转移酶,在医学和临床上具有研究和应用价值。
4 结论
本研究首次对深海真菌D. cejpii中GliG、GliI、GliO三个基因的启动子序列进行克隆,采用荧光素酶表达载体验证了GliG启动子的转录活性,并在酿酒酵母细胞中验证了GliG启动子具有转录表达潮霉素抗性基因的功能。本研究为后期通过对D. cejpii胶霉毒素生物合成基因进行转录调控和异源表达获得更多高活性的新型胶霉毒素奠定分子生物学基础。
[1] Gardiner DM, Waring P, Howlett BJ. The epipolythiodioxopiperazine(ETP)class of fungal toxins:distribution, mode of action functions and biosynthesis[J]. Microbiology, 2005, 151:1021-1032.
[2] Kwon-Chung KJ, Sugui JA. What do we know about the role of gliotoxin in the pathobiology ofAspergillus fumigatus[J]. Medical Mycology, 2009, 47:S97-S103.
[3] Duell ER, Glaser M, Chapelain CL, et al. Sequential inactivation of gliotoxin by the S-methyltransferase tmtA[J]. ACS Chemical Biology, 2016, 11(4):1082-1089.
[4] 周万青, 沈瀚, 张之峰, 等. 烟曲霉胶霉毒素的研究进展[J].中国真菌学杂志, 2011, 6(2):1673-3827.
[5] Dolan SK, O’Keeffe G, Jones GW, et al. Resistance is not futile:gliotoxin biosynthesis, functionality and utility[J]. Trends in Microbiology, 2015, 23(7):419-428.
[6] Corden J, Wasylyk B, Buchwalder A, et al. Promoter sequences of eukaryotic protein-coding genes[J]. Science, 1980, 209(4463):1406-1414.
[7] Even DY, Kedmi A, Ideses D, et al. Functional screening of core promoter activity[J]. Methods in Molecular Biology, 2017, 1651 :77-91.
[8]Punt PJ, Dingemanse MA, Kuyvenhoven A, et al. Functional elements in the promoter region of theAspergillus nidulansgpdA gene encoding glyceraldehyde-3-phosphate dehydrogenase[J].Gene, 1990, 93(1):101-109.
[9] 林涛, 黄建忠. 丝状真菌启动子研究进展[J]. 安徽农业科学,2013, 41(7):2862-2863, 2865.
[10] Fan Z, Sun ZH, Liu Z, et al. Dichotocejpins A-C :new diketopiperazines from a deep-sea-derived fungusDichotomomyces cejpiiFS110[J]. Marine Drugs, 2016, 14(9):164.
[11] 杨晓岚, 陈玉婵, 李浩华, 等. 23株海洋真菌的分子鉴定及其抗植物病原真菌和细胞毒活性的研究[J]. 生物技术通报,2014, 46(8):132-137.
[12] Quan JY, Tian JD. Circular polymerase extension cloning for high-through put cloning of complex and combinatorial DNA libraries[J]. Nature Protocols, 2011, 6(2):242-251.
[13] Ashrafi1 M, Fardsi M, Mirshamsi A, et al. Isolation and sequence analysis of gpdII promoter of the white button mushroom(Agaricus bisporus)from strains Holland737 and IM008[J]. International Journal of Horticultural Science and Technology, 2015, 2:33-41.
[14] Wu SX, Letchworth GJ. High efficiency transformation by electroporation ofPichia pastorispretreated with lithium acetate and dithiothreitol[J]. Drug Discovery and Genomic Technologies, 2004, 36:152-154.
[15] Punt PJ, Biezen NV, Conesa A, et al. Filamentous fungi as cell factories for heterologous protein production[J]. Trends in Biotechnology, 2002, 20(5):200-206.
[16] Helena Nevalainen KM, Te’o VSJ, Bergquist PL. Heterologous protein expression in filamentous fungi[J]. Trends in Biotechnology, 2005, 23(9):468-474.
猜你喜欢
环球时报(2022-09-20)2022-09-20
空间科学学报(2021年1期)2021-05-22
今日农业(2020年24期)2020-12-15
环境保护与循环经济(2017年5期)2018-01-22
中国调味品(2017年2期)2017-03-20
现代检验医学杂志(2016年5期)2016-08-20
中国果菜(2016年9期)2016-03-01
中国蔬菜(2015年9期)2015-12-21
中国科技信息(2015年2期)2015-11-16
小资CHIC!ELEGANCE(2015年15期)2015-09-01
