
杨树和柳树基因组共线性的可视化分析
2018-05-18 01:25徐逸卿杜思源蒋安纳王启昂薛倚鹭
江苏农业科学 2018年8期
徐逸卿,杜思源,蒋安纳,王启昂,薛倚鹭
(南京林业大学信息科学技术学院,江苏南京210037)
在杨柳科中,杨属和柳属是姐妹属[1],它们广泛地分布在北半球地区,具有适应力强、生长迅速、易繁殖、用途广等特点,是维护生态环境和解决木材短缺的重要物种[2]。杨属和柳属各自拥有相对较小的基因组,随着研究的大量开展以及基因组资源的迅速增长,杨柳科植物成为了木本植物在遗传学研究中的模式生物。在杨属中,毛果杨(Populus trichocarpa)的全基因组测序及染色体的组装早在2006年就已经完成[3],是第一个被测序的木本植物,之后杨属的物种陆续被测序;在柳属中,簸箕柳(Salix suchowensis)的全基因组测序、染色体组装分别于 2014、2016年完成[4-5]。细胞遗传学研究表明,杨属和柳属这2个属主要包含二倍体植物,其单倍型染色体数目是19对(n=19)[6]。在将杨树的基因组和柳树的遗传图谱进行比较时,研究人员经发现了染色体裂变、融合等现象[7]。但是遗传图谱与基因组相比,精度较低,因此需要通过对基因组信息进行分析来研究杨柳科的进化机制。
基因共线性是指在具有同源性关系的2个物种中,其基因组中有共同的连锁基因,且同源基因的相对顺序具有较高保守性的现象[8]。在生物进化中,基因组会在全基因组复制、染色体重组、染色体倒位和易位等过程中发生结构和数量的变化[9-10]。因此,基因组的共线性分析在非编码序列的确认[11]、新测序物种的注释[12]和全基因组复制事件的估计[8]等过程中具有重要作用。本研究使用 MCScanX[13]、VGSC[14]这2个软件分别进行共线性分析和作图,对杨树和柳树的种内、种间基因组同源关系进行分析,并绘制相应的关系图,进而探讨导致杨属和柳属进化的基因组机制。本研究结果可为研究杨柳科祖先基因组复制事件提供重要依据。
1 材料与方法
1.1 试验数据
本试验采用杨柳科的毛果杨和簸箕柳这2个物种进行基因组内和基因组间的共线性分析,从而确定杨树和柳树的进化机制。毛果杨是第一个被用于全基因组测序的树种,具有生长速度快、基因组较小、经济价值高等特点。2006年,Tuskan等利用鸟枪测序法完成毛果杨基因组的测序,结果表明,毛果杨基因组内含有碱基对4.85亿个,染色体19对,推测基因数量为45 555个[3]。随着国内外有关研究的大量开展,毛果杨现已成为木本植物研究中的模式物种[15]。毛果杨的全基因组信息可从JGI数据库[16]下载,下载地址为http://genome.jgi.doe.gov/pages/dynamicOrganismDownload.jsf?organism=Ptrichocarpa。由于大多数林木研究物种的世代周期较长,多为高大的乔木,实际操作麻烦,而柳属中的簸箕柳的世代周期只有1年,个体较小,取材方便,并且易于栽培,对环境要求不高,可以极大提高研究人员的试验效率,因此簸箕柳成为了木本植物研究的新热点。Dai等于2014年完成了簸箕柳的全基因组测序[4],共发现了26 599个编码基因,其中20 261个基因与杨树基因同源。柳树的全基因组信息发布的网址为 http://115.29.234.170/willow,可以直接下载。下载的数据包括全基因组的序列(Fasta格式,https://en.wikipedia.org/wiki/FASTA_format)和基因注释文件(GFF格式,www.gmod.org/wiki/GFF3),这些都被广泛应用于常见的基因组装软件和数据库。
1.2 试验工具软件
全基因组数据为科研人员提供了大量的信息,在全基因组水平上进行共线性分析是比较基因组学的重要研究内容。因此,越来越多研究共线性的方法被提出,早期的软件多采用传统的聚类算法,如 OrthoCluster[17-18]、ADHoRe[19]和 Maxgap Clusters by Multiple Sequence Comparison(简 称MCMuSeC)[20],对邻近的基因对进行匹配。这些软件在计算过程中由于影响因素较多,结果的可靠性并不是很高。另一种常见的算法是使用动态算法线性双向匹配基因对,并且利用“锚基因”对相邻的线性基因进行打分。此类软件包括ColinearScan[21]、MCScanX[8]和 SyMAP[12]等。ColinearScan利用基因组蛋白质的比对序列确定同源基因对是否存在,有效地预测了待测基因的共线性。SyMAP的优点在于寻找共线性基因的速度很快,但是由于参数的设置只能识别大片段的共线性基因,小片段的共线性基因很容易被漏掉。MCScanX(简称Multiple Collinearity Scan)是目前最流行的共线性分析软件,它采用匹配的锚点扫描多个基因中的序列或子序列,然后对假定的同源染色体区域进行匹配计分,最后给出计算的共线性分值。综上所述,本试验选用MCScanX软件作为共线性分析的主要工具。
上述共线性分析软件的侧重点集中在数据的处理上,缺少了对下游数据分析结果的可视化,绝大多数都没有提供可视化的输出接口。因此,近年来出现了一些专门进行共线性图形化的软件,例如 SynChro[22]、GSV[23]和 Easyfig[24],但是这些软件只能提供双线图的显示,显示效果不直观并且也不利于导出和发表。Circos是科学可视化领域一个著名的图形化工具,它可以将基因匹配与比较分析异同的结果用圆形图案表示出来,提供点阵图和矢量图的输出,在生物信息学研究中很受欢迎[25]。但它也仅限于圆形图形的处理,使得共线性研究受到了一定程度的限制。为了弥补这些不足,MCScanX设计并实现了15个用于共线性分析和显示的工具,并提供了点图、圆图、双线图等多种展示类型,实用性很强。然而,MCScanX只能允许用户在命令行的环境中进行操作,并且只能输出低分辨率的点阵图,在高通量测序逐渐普及的今天,成为了数据精细化分析的瓶颈。VGSC(A Web-Based Vector Graph Toolkit of Genome Synteny and Collinearity)[14]是 2016年最新发布的共线性分析作图的在线平台,自发布以来,该在线平台已经在生物学领域得到了广泛应用,不仅推动了生物进化分析的发展,也给基因家族分析、结构变异、新物种全基因组注释等方面带来了极大的便利。该平台最大的优势就在于分析结果的矢量图输出能力。如表1所示,经过比较,本试验选用VGSC绘制杨、柳物种基因组内与组间共线性关系图。

表1 共线性可视化软件基本信息
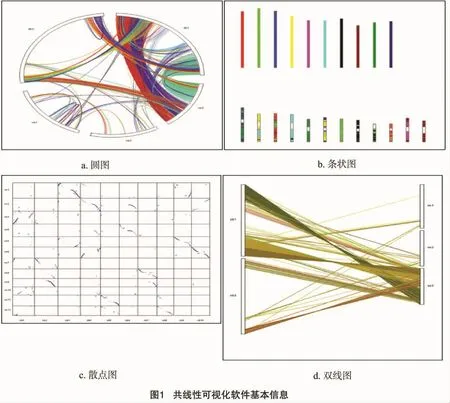
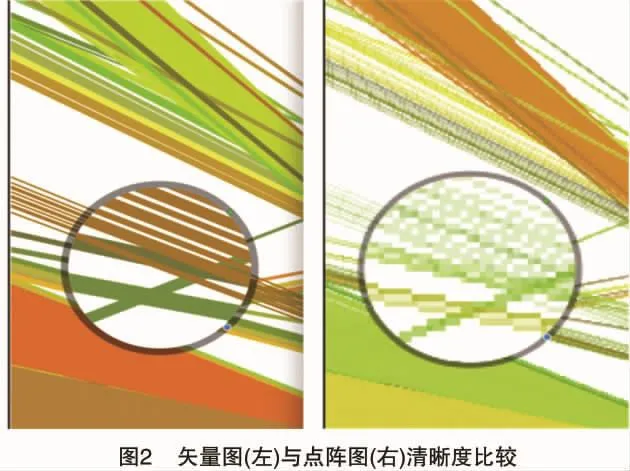
VGSC可以利用用户上传的GFF格式的基因注释文件和共线性分析生成的关系文件计算并生成对应的图形结果。如图1所示,VGSC提供了圆图、条状图、散点图和双线图4种不同的图形。用户可以根据研究的需要,选取并创建合适类型的图形,并且每种类型都支持高分辨率的点阵图(如BMP、JPEG和PNG)和高清晰度的矢量图的输出方式(如PDF、EPS和SVG)。VGSC的一个重要的特性就是它的矢量图输出能力,如图2演示了点阵图像和矢量图形在细节表现中的差异,随着高通量测序的不断发展,新测序技术带来的海量数据常常生成巨幅的运算结果。因此,在进行比对时,矢量图形的大小无关性和方向无关性的特点,对研究的开展及其报告的展示产生了积极的意义。
1.3 分析过程
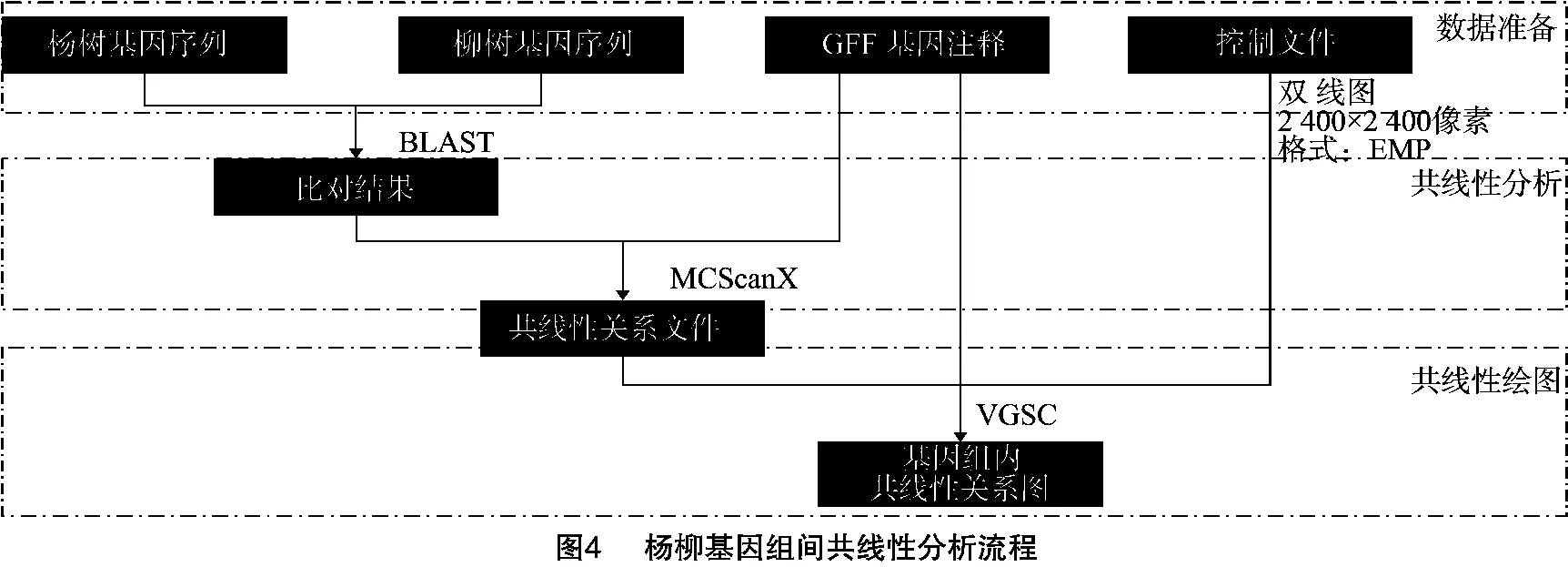
杨柳基因组内、基因组间的共线性分析流程分别如图3、图4所示。在进行基因组内共线性研究时,第1步是利用序列比对检索工具BLASTp,分别将该物种内的每条染色体与其他所有染色体进行蛋白质序列比对,得到各自的比对结果;在进行组间共线性分析时,对杨树基因组和柳树基因组进行1次全基因组比对检索。第2步是利用MCScanX计算比对结果的BLAST文件和GFF格式的基因注释进行计算,得到共线性分析结果collinearity文件。第3步是在VGSC的在线服务平台上传得到的collinearity文件和GFF格式的基因注释文件,配置参数并作图得到共线性图形,保存为矢量图形便于进一步分析。

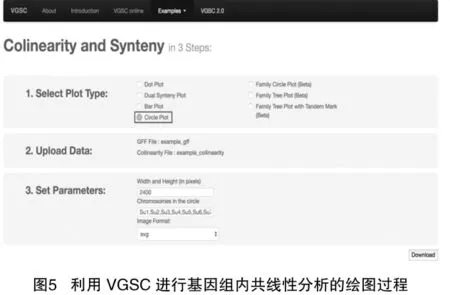
具体地讲,图5、图6分别演示了第3步VGSC对杨树、柳树基因组内和基因组间共线性作图的参数。VGSC的图形绘制流程与常见的图形化工具类似,首先,它提供了圆图、条状图、散点图和双线图4种不同的图形,其中圆图能够直观展现1条染色体和多条染色体之间的共线性关系,双线图则更适合展现2条染色体间的共线性关系。在进行基因组内和组间共线性绘图时,我们分别选取圆图、双线图作为输出图形。其次,上传BLASTp工具的比对结果和GFF基因注释文件。再次,对绘图参数进行配置,包括图形的大小、输出文件格式和共线性分析的染色体名称。
2 结果与分析
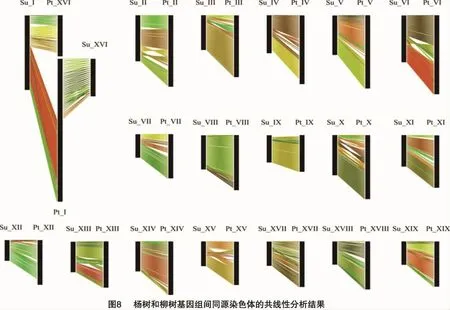
以上共线性分析及作图对于揭示杨柳科植物的近源关系具有积极的作用。通过观察杨树的共线性分析圆形图可以发现,各个染色体间同源片段的整体分布情况与Tuskan等的研究结果[3]大致一致,但是由于基因组信息的不断更新而造成了一些小差异(图7)。杨树基因组内的每条染色体都可以在其他染色体上找到同源片段,柳树基因组内也是如此。通过比较杨树和柳树对应的染色体的同源片段分布情况,我们可以发现只有Ⅰ、Ⅲ、Ⅵ和ⅩⅥ这4条染色体的同源关系图存在较大差异(图7),其余15条染色体几乎一样。通过试验还可以发现,杨树的1号染色体的上半部分的同源基因片段在其3号染色体上,而柳树的1号染色体的上半部分的同源基因片段在6号染色体上。杨树的16号染色体的同源基因片段在其6号染色上,而柳树16号染色体的同源基因片段基本上在3号染色体上。假设杨树和柳树基因组之间的差异是由1号染色体和16号染色体之间的重排引起的,为了验证此猜想,可以对杨树和柳树基因组中相应的染色体进行共线性分析比较。如图8所示,在对杨树和柳树基因组间共线性进行分析时,除了1号染色体和16号染色体这2组染色体间存在较大的差异,其他组染色体间的共线性关系都很高。而杨树1号染色体与柳树16号染色体,以及柳树1号染色体与杨树16号染色体共线性分析结果显示,杨树1号染色体的上半部分与柳树的16号染色体同源,而下半部分与柳树的1号染色体同源。杨树16号染色体的同源片段存在于柳树1号染色体的上半部分。这一发现为发生在1号染色体和16号染色体间的断裂融合提供了有力证据。除此之外,杨树和柳树基因组内和基因组间的共线性分析结果表明,还存在一些染色体间的重排现象。Hou等采用简单序列重复(simple sequence repeats,简称SSR)标记分析进一步验证了杨树、柳树的1号染色体与16号染色体间发生了重排[5]。

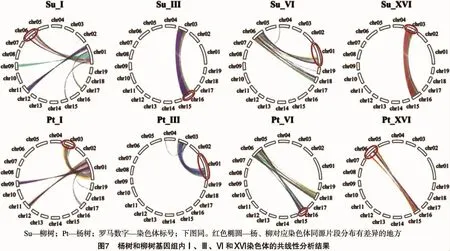
3 讨论与结论
共线性分析软件的出现极大地推动了基因共线性分析的发展,近年来,更多的工具能够让用户以在线形式快速高效地将共线性分析结果以高清晰度的矢量图或高质量点阵图的方式输出。通过比对分析VGSC绘制的杨树、柳树基因组内和基因组间共线性关系图,笔者发现基因组内不同染色体间存在大量重复片段,并且基因组间大多数染色体间的共线性关系很高。这一现象说明,杨树和柳树的分化是在古四倍体基因组二倍化之后完成的。经过基因组间的共线性分析,笔者发现了2个大的染色体重排现象,由此推测1号染色体与16号染色体间发生了断裂融合,这一结果说明杨树和柳树没有同时进行分化,而是一个物种是由另一个物种进化而来的。Dorn等研究表明,杨树的进化早于柳树,因此可以推测杨树向柳树进化的过程:古四倍体祖先的基因组在染色体的断裂融合之后进行了二倍化,由此出现了杨树[26-27]。杨树在进化的过程中,其1号染色体和16号染色体间发生了重排事件,进而出现了现代柳树的祖先。综上所述,本研究结合了MCScanX的分析工具和VGSC的高质量图形化服务,更直观地再现了杨树和柳树基因组内和基因组间染色体基因的对应关系,推测了杨属和柳属进化的机制,为杨柳科植物的起源提供了重要依据,为更好地认识杨柳科植物打下了基础。
参考文献:
[1]Heywood V H,Moore D M,Richardson IB K,et al.Flowering plants of the world[M].Oxford:Oxford University Press,1993:316.
[2]Isebrands JG,Richardson J.21st session of the international poplar commission(IPC 2000).Poplar and willow culture:meeting the needs of society and the environment[J].Art Education,2000,41(1):9-17.
[3]Tuskan G A,di Fazio S,Jansson S,et al.Supporting online material for the genome of black cottonwood,Populus trichocarpa(Torr.&Gray)[J].Science,2006,313(5793):1596-1604.
[4]Dai X,Hu Q,CaiQ,etal.Thewillow genome and divergentevolution from poplar after the common genome duplication[J].Cell Research,2014,24(10):1274-1277.
[5]Hou J,Ye N,Dong Z,et al.Major chromosomal rearrangements distinguish willow and poplar after the ancestral“Salicoid”genome duplication[J].Genome Biology&Evolution,2016,8(6):1868-1875.
[6]Blackburn K B,Harrison JW H.A preliminary account of the chromosomes and chromosome behaviour in the salicaceae[J].Annals of Botany,1924,38(150):361-378.
[7]Berlin S,Lagercrantz U,Arnold S V,et al.High-density linkage mapping and evolution of paralogs and orthologs in Salix and Populus[J].BMC Genomics,2010,11(1):129.
[8]Tang H,Bowers JE,Wang X,et al.Synteny and collinearity in plant genomes[J].Science,2008,320(5875):486-488.
[9]Dujon B,Sherman D,Fischer G,et al.Genome evolution in yeasts[J].Nature,2004,430(6995):35-44.
[10]Nakatani Y,Takeda H,Kohara Y,et al.Reconstruction of the vertebrate ancestral genome reveals dynamic genome reorganization in early vertebrates[M].Japan:Springer,2011.
[11]Lyons E,Pedersen B,Kane J,et al.Finding and comparing syntenic regions among Arabidopsis and the outgroups papaya,poplar,and grape:CoGe with rosids[J].Plant Physiology,2008,148(4):1772-1781.
[12]Soderlund C,Bomhoff M,Nelson W M.SyMAP v3.4:a turnkey synteny system with application to plant genomes[J].Nucleic Acids Research,2011,39(10):e68.
[13]Wang Y,Tang H,Debarry J D,et al.MCScanX:a toolkit for detection and evolutionary analysis of gene synteny and collinearity[J].Nucleic Acids Research,2012,40(7):e49.
[14]Xu Y,Bi C,Wu G,et al.VGSC:A web-based vector graph toolkit of genome synteny and collinearity[J]. Biomed Research International,2016,2016(1):7823429.
[15]张 勇,张守攻,齐力旺,等.杨树——林木基因组学研究的模式物种[J].植物学报,2006,23(3):286-293.
[16]Grigoriev IV,Nordberg H,Shabalov I,et al.The genome portal of the Departmentof Energy JointGenome Institute[J].Nucleic Acids Research,2012,40:D26-D32.
[17]Vergara IA,Chen N.Using OrthoCluster for the detection ofsynteny blocks among multiple genomes[M]//Current Protocols in Bioinformatics.John Wiley&Sons Inc,2009.
[18]Zeng X,Nesbitt M J,Pei J,etal.OrthoCluster:a new tool formining synteny blocks and applications in comparative genomics[C]//Proceedings of the International Conference on Extending Database TechnologyAdvances in Database Technology,2008:656-667.
[19]Vandepoele K,Saeys Y,Simillion C,et al.The automatic detection of homologous regions(ADHoRe) and its application to microcolinearity between Arabidopsis and rice[J]. Genome Research,2002,12(11):1792.
[20]Ling X,He X,Xin D.Detecting gene clusters under evolutionary constraint in a large number of genomes[J].Bioinformatics,2009,25(5):571-577.
[21]Wang X,Shi X,Li Z,et al.Statistical inference of chromosomal homology based on gene colinearity and applications to Arabidopsis and rice[J].BMC Bioinformatics,2006,7(1):447.
[22]Drillon G,Carbone A,Fischer G.SynChro:a fast and easy tool to reconstruct and visualize synteny blocks along eukaryotic chromosomes[J].PLoSOne,2014,9(3):e92621.
[23]Revanna K V,Chiu C C,Bierschank E,et al.GSV:a web-based genome synteny viewer for customized data[J]. BMC Bioinformatics,2011,12(1):316.
[24]Sullivan M J,Petty N K,Beatson SA.Easyfig:a genome comparison visualizer[J].Bioinformatics,2011,27(7):1009-1010.
[25]Gascoyne R D,Krzywinski M,Birol I,et al.Circos:an information aesthetic for comparative genomics[J].2009,
[26]Dorn R D.A synopsis of American Salix[J].Canadian Journal of Botany,2011,54(24):2769-2789.
[27]Skvortsov A K. Willows of Russia and adjacent countries:taxonomical and geographical revision[M].Joensuu:University of Joensuu Press,1999:1-307.
猜你喜欢
华人时刊(2023年1期)2023-03-14
汉字汉语研究(2021年2期)2021-08-30
科学与财富(2021年3期)2021-03-08
汉字汉语研究(2019年2期)2019-08-27
——以多重共线性内容为例
长沙航空职业技术学院学报(2019年2期)2019-07-13
温州大学学报(自然科学版)(2019年2期)2019-06-04
少儿科学周刊·儿童版(2017年5期)2017-06-29
河北书画研究(2016年3期)2016-04-28
测绘学报(2013年1期)2013-01-11
