
全外显子组测序证实家族性肺动脉高压骨形成蛋白受体2基因新突变的研究
2018-05-21 08:34郑亚国林松张航谢渡江周陵陈绍良
中国循环杂志 2018年5期
郑亚国,林松,张航,谢渡江,周陵,陈绍良
肺动脉高压(pulmonary arterial hypertension,PAH)是一种以肺动脉内膜增生、中膜肥厚及丛样病变为特征的一种疾病[1]。PAH的发病机制与遗传因素密切相关,目前指南把可遗传性PAH单独列为PAH的1个亚型。已知骨形成蛋白受体2(bone morphogenetic protein receptor 2, BMPR2)、活化素受体 类 激 酶 1(activin receptor-like kinase 1, ALK1)、Smad蛋 白 9(SMAD9)、 小 窝 蛋 白 l(caveolin-1,CAV-l)和钾离子通道蛋白3(KCNK3)等多种基因突变和PAH发病有关[2]。目前研究发现,BMPR2是目前已知的最主要的PAH致病基因,约70%的可遗传性PAH及26%的特发性PAH患者存在BMPR2基因突变,公共数据库ClinVar(https://www.ncbi.nlm.nih.gov/clinvar)共计报道了超过500个BMPR2的不同突变位点[3-5]。本研究中,我们利用全外显子组测序方法对一个PAH家系进行研究,以发现BMPR2基因的突变位点。
1 资料与方法
研究对象:收集2016-11来本院就诊的一个中国汉族PAH家系的资料, 3代共8人,所有成员均为江苏籍汉族人,有2位已确诊为PAH,其中男性1例(先证者),女性1例,为母子关系(图1)。另选择100名健康志愿者作为正常对照。PAH的诊断标准依据2015年欧洲心脏病学会PAH诊治指南[1]。对所有家系成员进行病史采集、体格检查、心电图及超声心动图检查,同时对先证者及超声心动图怀疑PAH的家系成员进行生化、免疫学检查、六分钟步行距离及右心导管检查等更为详尽的临床评估。所有入选家系成员和健康志愿者均签署知情同意书。
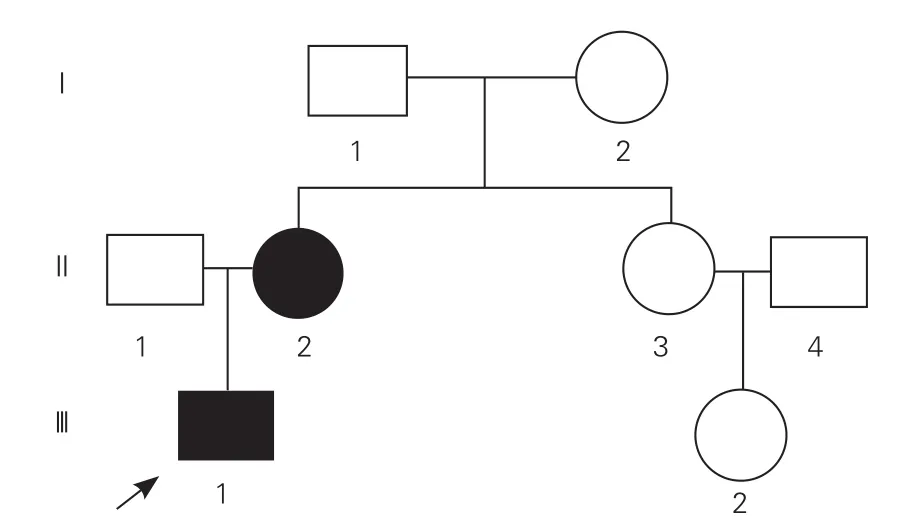
图1 该肺动脉高压家系图
标本采集及DNA提取:选用乙二胺四乙酸(EDTA)抗凝管分别收集本研究家系8名成员的外周静脉血5 ml。按照DNA提取试剂盒(Qiagen, 德国)说明书提取基因组DNA。采用紫外分光光度法测定提取DNA的浓度与纯度,-20℃分装保存。
全外显子组测序:先证者及其母亲与先证者的父亲及姨妈(正常者)作对照,进行外显子测序分析(诺禾致源,北京)。将基因组DNA经Covaris破碎仪随机打断成长度为180~280 bp的片段,经末端修复和加A尾后在片段两端分别连接上接头制备DNA文库。带有特异index的文库pooling后与生物素标记的探针进行液相杂交,再使用带链霉素的磁珠将基因上的外显子捕获下来,经聚合酶链式反应(PCR)线性扩增后进行文库质检,合格即可运用Illumina平台进行测序。
生物信息学分析:获得原始测序序列后,在有参考序列或参考基因组(GRCh37/hg19)的情况下,进行信息分析流程,大致包括以下三个部分:测序数据质量评估,变异检测,变异筛选及与疾病相关性的预测。同时,对数据进行质控检测,包括测序深度、覆盖度均一性等分析。序列比对使用Burrows-Wheeler Aligner 软件。在比对结果的基础上,我们利用SAMtools识别SNP和InDel位点,并采用国际惯用的过滤标准对这些位点进行过滤。测序结果经公共遗传数据库dbSNP138及千人基因数据库过滤,去掉公共人群单核苷酸多态性突变和同义突变。剩余的突变根据ACMG开发的针对序列变异解读的标准和指南对突变位点有害性进行分类,留取致病性的变异位点及可能致病性的变异位点进行分析。再取2例患者突变的交集。
致病基因家系样本测序验证:将数据分析后得到的可能的致病基因BMPR2的突变位点,设计引物,在其余4个家系成员及100名健康志愿者中进行Sanger测序验证。采用Primer 5软件设计引物,用PCR技术扩增BMPR2基因的6号外显子,引物由上海生工公司合成,引物序列为:BMPR2-EX6F:AAACACTTGCAGCTGATTGG;BMPR2-EX6R:TTACATTGGGATAGTACTCCATCAC。DNA样 本经PCR扩增后在自动测序仪ABI 3730XL Genetic Analyzer(赛墨飞,美国)上进行测序。
2 结果
临床资料:先证者为14岁男性,因为活动后胸闷、气喘先后在三家医院就诊,查体发现肺动脉第二心音亢进,心电图示电轴右偏,肺性P波,右心室重度肥厚,超声心动图示右心房、右心室扩大,右心室壁增厚,室间隔左移,左心室呈“D”型,估测肺动脉收缩压91 mmHg(1 mmHg=0.133 kPa);右心导管测量右心房压11 mmHg,肺动脉收缩压/舒张压119/56 mmHg,肺动脉平均压77 mmHg,肺毛细血管锲压11 mmHg,心输出量4.92 L/min,肺小动脉阻力13.4 Wood units,急性肺血管扩张试验阴性,确诊PAH后先后给予波生坦(125 mg,bid)、他达拉非(10 mg,qd)及贝前列素钠片(60 μg,tid)治疗。另一位确诊的PAH患者为先证者母亲,38岁,活动后胸闷、气喘10年,目前活动耐力重度受限,心电图示频发室性早搏,电轴右偏,肺性P波,右心室重度肥厚,超声心动图示右心房、右心室扩大,估测肺动脉收缩压132 mmHg,随访至2017-03,患者母亲死于右心衰竭。
全基因组外显子测序结果:家系中2例患者(Ⅱ2,Ⅲ1)及两名正常者(Ⅱ1,Ⅱ3)的外显子测序结果见表1。我们在2例患者的编码区发现了23 302及23 021个SNPs位点及629和582个编码Indels。我们重点关注非同义突变位点、变异性剪接位点、插人和缺失的移码突变位点,并排除在dbSNPl38数据库及1 000 Genome Project数据库中存在的突变位点。同时我们取2例患者的交集,经过一系列筛选后剩下127个突变位点,包括114个SNPs和13个Indels。我们根据得到的结果,与已报道的遗传学PAH相关基因数据库中比对,发现在此家系中发现BMPR2基因6号外显子747位点的移码突变(BMPR2:NM_001204:e xon6:c.747_748insCCTTTGATGGAACATGA:p.V250fs)在2例PAH患者中均存在,在2名对照者中均不存在,很可能是该家系的致病基因。家系中的2例患者并不携带其他已知的与PAH相关的致病基因,如 CAV-1、ALK1、KCNK3、SMAD9、BMPR1B、ENG、MADH9及PPHR等。
BMPR2突变位点家系验证及扩大样本验证:为了确定BMPR2移码突变(hg19 chr2: g.203383670G>GCCTTTGATGGAACATGA)是该家系的致病基因,我们设计了针对该突变位点的引物。并用Sanger测序法对该家系8个成员的DNA样本进行测序验证。测序结果与人类基因组数据库进行比对分析,发现2例患者均携带有BMPR2插入杂合突变,而家族其余成员均没有该位点插入或缺失突变(图2)。BMPR2插入杂合突变(c.747_748.insCCTTTGATGGAACATGA)在该家系中表现出与临床表型的完全共分离。
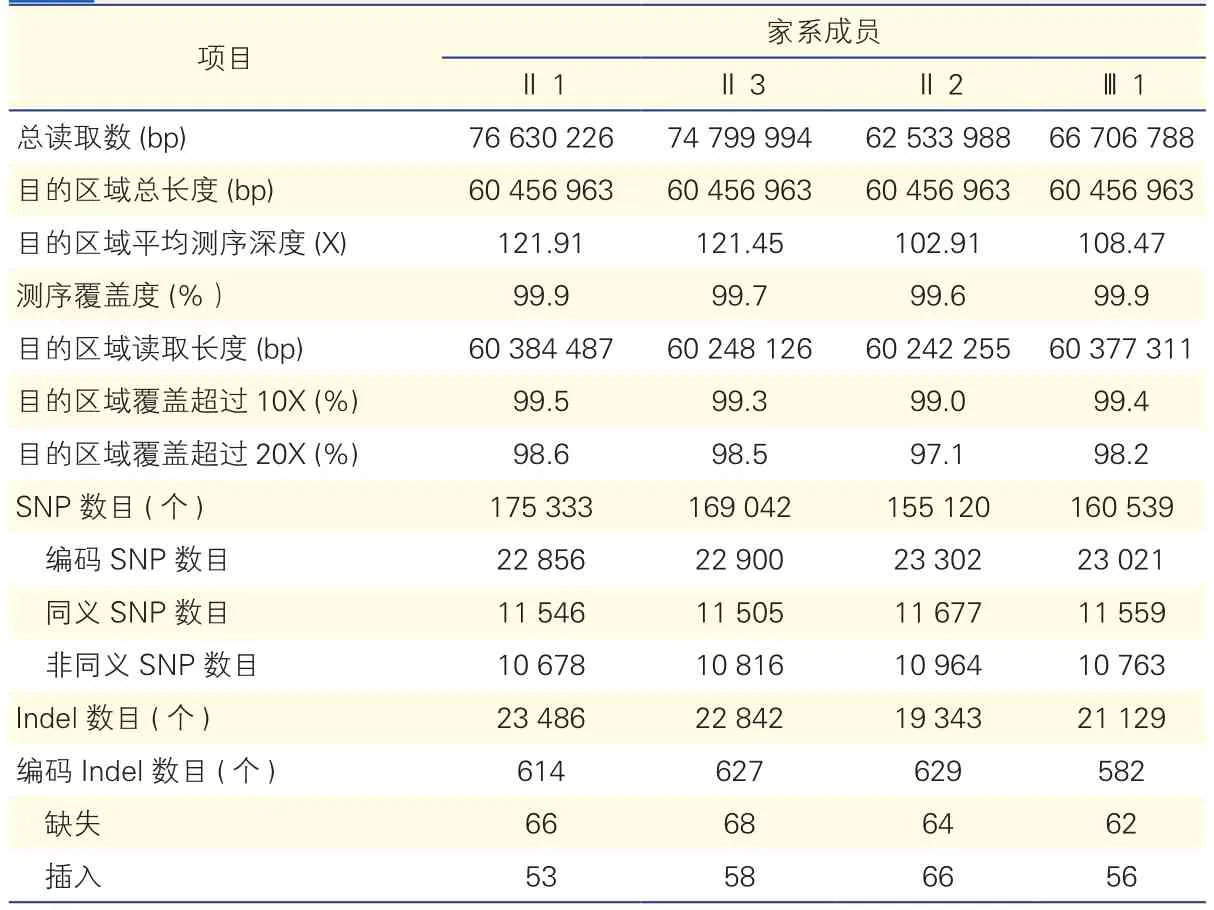
表1 外显子测序数据
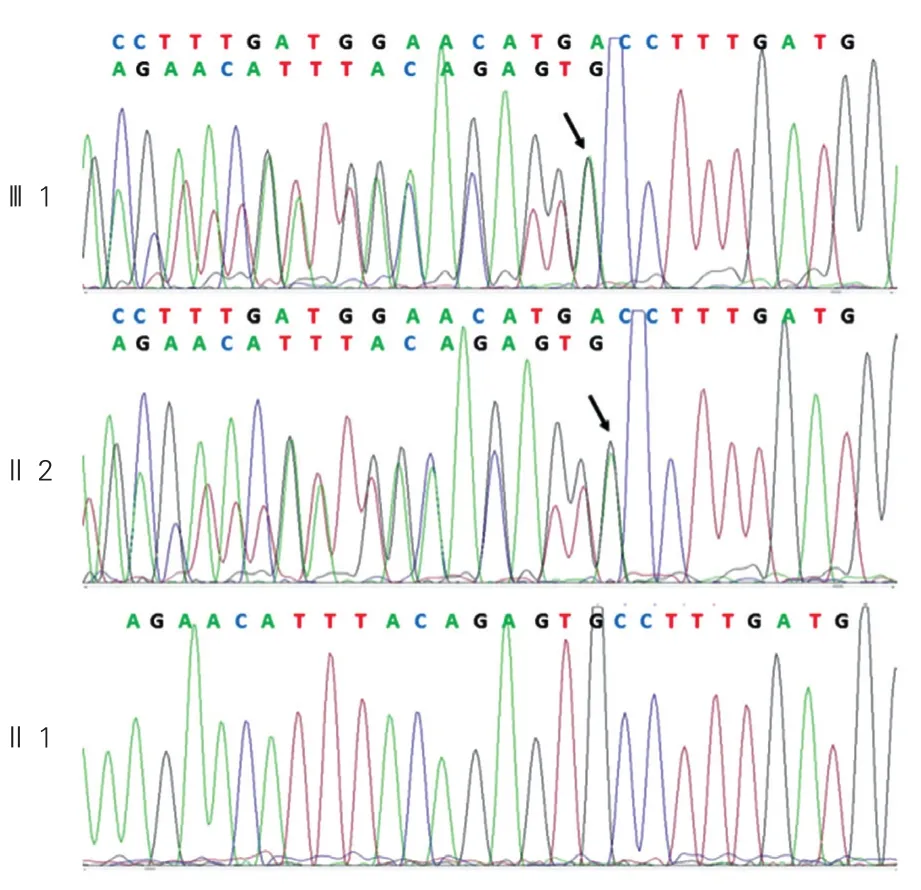
图2 骨形成蛋白受体2基因突变位点DNA序列图
我们进一步在100名健康志愿者样本中进行验证,未发现有BMPR2基因6号外显子747位点的插入杂合突变。通过CADD软件分析,该突变致BMPR2的250~256位氨基酸发生移码突变(p.V250fs),并自257位终止;插入突变破坏了读码框,翻译截短蛋白,从而造成BMPR2蛋白的结构和功能缺陷。因此,BMPR2基因6号外显子747位点的插入杂合突变(c.747_748.insCCTTTGATGGAACATGA)是造成该家系患者肺动脉高压的遗传学病因。
3 讨论
目前认为BMPR2基因突变引起BMPR2的功能缺陷是PAH的重要发病机制。BMPR2基因突变与PAH患者的临床表型及预后密切相关。携带BMPR2基因突变的患者临床表型更为严重,发病时间更早,血液动力学改变更严重,急性肺血管扩张试验的阳性率更低,预后更差[6,7]。
在本研究中,我们利用高通量的二代测序技术对患者样本进行全外显子组测序。全外显子测序利用目标序列捕获技术将基因组的全部外显子区域DNA捕获后进行高通量测序,不仅能快速发现罕见遗传病的致病基因,也能用于多基因引起的常见疾病,从而揭示这些疾病的遗传致病机理。目前全外显子测序用于肺动脉高压的研究发现许多和肺动脉高压发病有关的新基因[8-10]。本研究中,我们在一个中国汉族PAH家系中发现了BMPR2基因6号外显子747位点新的插入杂合突变位点(c.747_748.insCCTTTGATGGAACATGA)。目前6号外显子上的已经报道的和肺动脉高压相关的突变包括无义突变(c.631C>T)、错义突变(c.604A>T、c.794A>G、c.797G>C 及 c.806G>T)、剪接突变(c.852+1G>C、c.853-1G>C) 及 移 码 突 变(c.612delA、c.690-691delAGinsT 及 c.673-679delCGTCCAG)[4,5]。
在此家系中,我们认为,BMPR2基因插入杂合突变(c.747_748.insCCTTTGATGGAACATGA)导致PAH表型,主要基于以下证据:(1)该突变经过外显子测序发现同时经过Sanger测序验证,并且在100名健康志愿者和dbSNP l38、千人基因数据库中都未发现有该突变;(2)该突变在本家系中表现出明显的遗传共分离现象,呈现常染色显性遗传特征,携带BMPR2基因突变的PAH患者是杂合子,等位基因中有一个是突变型,另一个是野生型;这与文献报道的PAH遗传特性一致;(3)BMPR2基因是一个目前公认的PAH发病相关的基因,该突变位点附近的很多突变位点已经被相关文献报道[4,5]。(4)BMPR2突变致BMPR2 250位缬氨酸后发生移码突变(p.V250fs),插入突变破坏了读码框,翻译截短蛋白,从而造成BMPR2蛋白的结构和功能缺陷。因此,我们认为该突变导致了PAH的发生。
该研究对于有家族史的患者,未常规使用一代测序排查常见致病基因突变,直接使用二代测序,最终发现的还是BMPR2基因上的突变,在一定程度上增加了科研经费支出。本研究报道了一个中国汉族人PAH家系,通过全外显子组测序技术发现了BMPR2基因的一个新的移码突变位点(c.747_748.insCCTTTGATGGAACATGA)。该位点在该家系中表现出共性分离,是该家系PAH发生的遗传学病因。本研究丰富了BMPR2基因的致病突变位点谱。
参考文献
[1] Galie N, Humbert M, Vachiery J, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension[J]. Eur Heart J,2015, 37(1): 67-119. DOI: 10. 1093/eurheartj/ehv317.
[2] 曾绮娴, 熊长明. 肺动脉高压遗传基因研究进展 [J]. 中华医学杂志, 2017, 97(8): 632-634. DOI: 10. 3760/cma. j. issn. 0376-2491.2017. 08. 016.
[3] 赵林, 袁安慧, 王晓建, 等. 肺动脉高压遗传修饰因素的研究进展[J]. 中华心血管病杂志, 2016, 44(9): 817-820. DOI: 10. 3760/cma. j.issn. 0253-3758. 2016. 09. 020.
[4] Machado RD, Southgate L, Eichstaedt CA, et al. Pulmonary arterial hypertension: a current perspective on established and emerging molecular genetic defects[J]. Hum Mutat, 2015, 36(12): 1113-1127.DOI: 10. 1002/humu. 22904.
[5] Machado RD, Eickelberg O, Elliott CG, et al. Genetics and genomics of pulmonary arterial hypertension[J]. J Am Coll Cardiol, 2009, 54(1):S32-42. DOI: 10. 1016/j. jacc. 2009. 04. 015.
[6] Evans JD, Girerd B, Montani D, et al. BMPR2 mutations and survival in pulmonary arterial hypertension: an individual participant data meta-analysis[J]. Lancet Respir Med, 2016, 4(2): 129-137. DOI: 10.1016/S2213-2600(15)00544-5.
[7] Elliott CG, Glissmeyer EW, Havlena GT, et al. Relationship of BMPR2 mutations to vasoreactivity in pulmonary arterial hypertension[J].Circulation, 2006, 113(21): 2509-2515. DOI: 10. 1161/CIRCULATIONAHA. 105. 601930.
[8] Ma L, Roman-Campos D, Austin ED, et al. A novel channelopathy in pulmonary arterial hypertension[J]. N Engl J Med, 2013, 369(4): 351-361. DOI: 10. 1056/NEJMoa1211097.
[9] Eyries M, Montani D, Girerd B, et al. EIF2AK4 mutations cause pulmonary veno-occlusive disease, a recessive form of pulmonary hypertension[J]. Nat Genet, 2014, 46(1): 65-69. DOI: 10. 1038/ng.2844.
[10] 张建华, 许小毛, 邹丽辉, 等. OR2T3基因与动脉性肺动脉高压的关联性分析[J]. 中华医学杂志, 2016, 96(16): 1256-1260. DOI: 10.3760/cma. j. issn. 0376-2491. 2016. 16. 007.
猜你喜欢
广西林业科学(2022年6期)2023-01-16
电子科技大学学报(2022年5期)2022-10-29
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中国生殖健康(2020年4期)2021-01-18
中国生殖健康(2020年2期)2021-01-18
中国生殖健康(2018年4期)2018-11-06
小学生导刊(2018年13期)2018-06-29
森林工程(2018年1期)2018-05-14
中国生殖健康(2018年2期)2018-01-12
