
一显性营养不良型大疱性表皮松懈症家系COL7A1基因突变分析
2018-05-30 00:47黄艳梅董双双曹靓磊郭利伟张咏鹤杨保胜
郑州大学学报(医学版) 2018年3期
黄艳梅, 董双双,曹靓磊,郭利伟,张咏鹤,李 茜,杨保胜
新乡医学院法医学院法医物证学教研室 河南新乡 453003
营养不良型大疱性表皮松懈症(dystrophic epidermolysis bullosa,DEB)是一种遗传异质性皮肤病,临床特征主要表现为皮肤黏膜对机械性损害易感并形成大疱,遗传方式以常染色体显性或隐性遗传。目前认为DEB的发病机制与编码基底膜下带锚原纤维的主要成分——Ⅶ型胶原的COL7A1基因突变有关,且具有遗传异质性[1-2]。本研究对一常染色体显性遗传DEB家系的COL7A1基因突变进行了分析,并对其发病的分子基础与临床表型之间的关系进行探讨。
1 对象与方法
1.1研究对象此家系为河南汉族人,无近亲结婚史,家系三代中,每一代均有患者,根据系谱图(图1)分析符合常染色体显性遗传。先证者男(Ⅲ1),2岁,出生后不久受到轻微外伤或摩擦后出现水疱、糜烂及破溃,主要集中在胫前、踝和上肢伸肌侧,愈后留有萎缩性瘢痕和粟丘疹,轻度瘙痒并且伴有指(趾)甲萎缩,甚至脱落。病情随年龄增长逐渐减轻。家系中其他患者均在出生或出生后不久轻度外伤后发生大疱性损害,愈后留有色素改变、粟丘疹及萎缩性瘢痕。成年后上述症状减轻,四肢伸肌侧形成苔藓样斑块及条索样改变。所有患者手、足甲板均有不同程度的增厚变形,均无黏膜和毛发改变。结合临床、组织病理学和电镜观察结果,确诊此家系为常染色体显性遗传型DEB。

箭头所指为先证者图1 DEB家系图
1.2全基因组DNA的提取在知情同意情况下,采集该家系成员10人外周静脉血各5 mL, 另取50名健康人外周静脉血作为对照。20 g/L EDTA 抗凝,然后采用有机酚-氯仿法提取血样基因DNA。
1.3PCR扩增及测序参照文献[3-4]合成72对引物,对COL7A1基因的118个外显子、内含子全部进行扩增并测序。其中13号外显子的引物序列为:正义5’-CCTTCTCACTCTGCGTCCCT-3’,反义5’-AACCAGGACCAGAGTGAGGC-3’; 63号外显子的引物序列为:正义5’-CTCCCAAAGTCCTTGAAATC-3’,反义5’-AGAACTATGAAGCCCAGCAC-3’;64号外显子的引物序列为:正义5’- AGTGGTTGGGTGCT GGGCTT-3’,反义5’-CACGTTCGCCCTGATGGAAA-3’;73号外显子的引物序列为:正义5’-GGGTG TAGCTGTACAGCCAC-3’,反义5’-CCCTCTTCCCT CACTCTCCT-3’ 。 PCR扩增体系为25 μL。PCR反应条件:95 ℃预变性2 min;94℃变性45 s,退火45 s,72 ℃延伸45 s,共33个循环;72 ℃再延伸10 min;4 ℃保存。扩增后经60 g/L聚丙烯酰胺凝胶电泳检测扩增产物,由美国Life Technology公司[英潍捷基(上海)贸易公司]进行双向测序。分析测序结果并与健康人及GenBank数据库中序列进行比较。
1.4新突变的致病性鉴定通过与NCBI dbSNP (http://www.ncbi.nlm.nih.gov/snp)、HGMD数据库(http://www.hgmd. cf.ac.uk /ac/all.php)和近5 a文献进行检索,证实检测到的突变是否为新发突变;利用Clustal X软件,对人、小家鼠、褐家鼠、牛、黑猩猩、猕猴、非洲爪蟾、斑马鱼等10个物种该位点的氨基酸进行保守性分析,根据保守性的强弱来判断突变致病性的大小;利用SIFT、Mutationtaster和PolyPhen软件对突变的危害性进行预测,以此判断突变的致病性。
2 结果
2.1PCR扩增结果所有72对引物在各自的条件下分别扩增各自的产物,包括所有118个外显子的编码序列以及侧翼内含子序列。PCR扩增产物所扩增的所有外显子通过凝胶电泳检测显示片段长度与预期的一致。
2.2分子遗传学检测结果对该DEB家系所有成员的COL7A1基因进行了基因测序,突变测序结果与50名健康人及GenBank 数据库(http://www. ncbi. nlm.nih. gov/genbank)进行对比分析,共筛选出5个突变位点,其中包括1个错义突变、3个同义突变和1个插入突变。见表1、图2。该家系中患者(Ⅰ1、Ⅱ1、Ⅲ1)均出现了这些突变,且均表现为杂合子。除此之外,先证者Ⅲ1第13号外显子存在c.1731_1732insA插入突变,导致了提前终止密码(PTC)的产生。在作为对照的50名健康人以及该家系表现正常者中未检测到上述突变。

表1 显性遗传型DEB COL7A1基因突变位点
2.3新突变的致病性鉴定
2.3.1 突变位点氨基酸的保守性分析结果 COL7A1肽链即α3链第2 006位氨基酸(即突变所在位置)在10个不同来源的跨物种中均为甘氨酸,说明该位点在进化过程中高度保守,该位点一旦发生突变,势必影响COL7A1蛋白的氨基酸组成,影响其螺旋结构,进而影响其正常功能。由此可以推断,本研究检测到的p.G2006S突变可能是致病性突变。
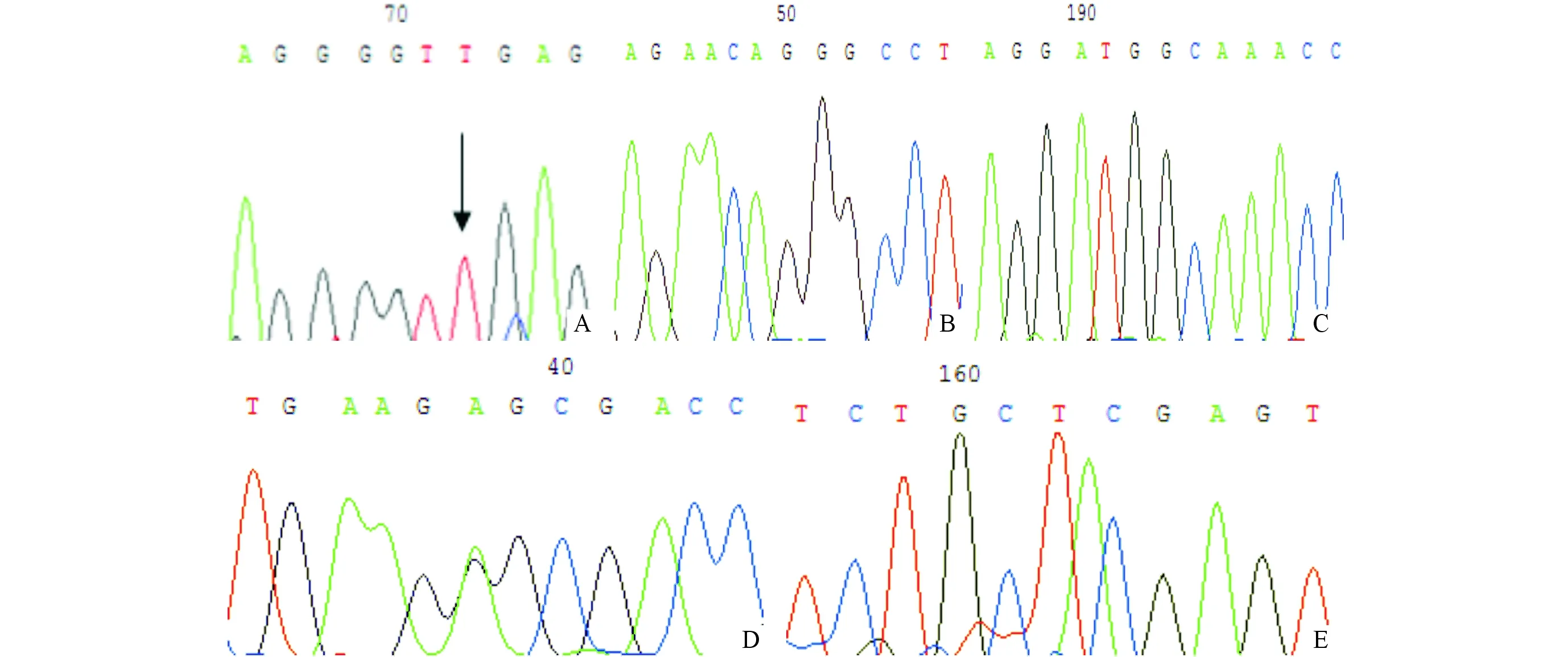
A:患者第1 641位碱基发生G→T突变,导致第547位色氨酸(TGG)突变为亮氨酸(TTG),即W547L;B:患者第5 451位碱基发生A→G 突变,导致第1 817位谷氨酰胺由(CAA)突变为(CAG);C:患者第5 508位碱基发生G→C突变,导致第1 836位甘氨酸由(GGG)突变为(GGC);D:患者第6 016位碱基发生G→A 突变,导致第2 006位甘氨酸(GGC)突变为丝氨酸(AGC),即G2006S ; E:先证者第1 731和第1 732位碱基之间插入了一个A碱基,导致PTC的产生,即c.1731_1732insA 图2 COL7A1基因测序结果
2.3.2 新发突变致病性的预测结果 Mutationtaster软件对插入突变c.1731_1732insA的预测结果为“可能影响蛋白质功能”;PolyPhen软件对错义突变p.G2006S的预测结果为“很可能致病”, Mutationtaster和SIFT软件预测结果为“可能影响蛋白质功能”,可见p. G2006S位的甘氨酸被丝氨酸替代是有害的,进一步证实错义突变p.G2006S突变可能是一种致病性突变。
3 讨论
DEB是一种少见的遗传性皮肤病,按其遗传形式可分为常染色体隐性和显性遗传[5-6]。DEB的发生与编码Ⅶ型胶原蛋白的COL7A1 基因突变有关。目前已知COL7A1 基因定位于人3p21.3区域,长度为23 000 bp[7]。 COL7A1基因在维持真表皮连接结构稳定性中具有非常重要的作用,它的突变会导致Ⅶ型胶原数量减少或形态异常,使得皮肤基底膜致密板下的锚原纤维(其主要成分是Ⅶ型胶原)不能维持正常的功能。皮肤在机械性外伤或表皮摩擦时,易发生表皮与真皮的分离,形成大疱[8-10]。COL7A1突变有多种。其中,位于三螺旋区的甘氨酸替代突变为DEB最常见的基因突变类型。由于Ⅶ型胶原中心三螺旋区的结构特征是Gly-Xaa-Yaa连续重复,胶原区Gly-Xaa-Yaa结构中甘氨酸是最小的氨基酸,对于胶原蛋白的结构和功能至关重要[11-12],当甘氨酸被其他种类的氨基酸替代后,影响Ⅶ型胶原的聚集,导致锚原纤维的改变,导致DEB的发生。
本研究发现了一个尚未见报道的插入突变c.1731_1732insA,即在COL7A1基因的第13号外显子上插入了一个A碱基,导致第13号外显子发生移码突变。本研究发现该突变造成编码区阅读框架的移位,产生提前终止密码(PTC),导致肽链合成提前终止,最终产生无意义的蛋白质[4]。该突变仅出现于先证者(Ⅲ1)COL7A1基因上,先证者在该家系患者中病情最严重,此突变是否与DEB临床表型有关还有待于进一步研究。
本研究亦发现了1个错义突变即c.6016G→A (p.G2006S)。该家系患者均出现此突变,表现为杂合子。先证者母亲(Ⅱ2)无DEB临床症状,基因检测结果未发现异常,50名健康人未检测到此突变,推测上述突变为该家系的致病突变位点。错义突变G2006S国外已被Mallipeddi等报道[13],但是本文患者病情与Mallipeddi等的报道不甚相同。由此可见,即使同一突变,在不同家系中可能表现为不同的表型,甚至在同一家系具有相同基因突变的成员临床表现也可能有较大差异。除此之外,作者在该家系的所有患者中还检测到了3个尚未报道的同义突变,分别为c.1641G→T、c.5451A→G和c.5508G→C。同义突变可能使原先的密码子变成稀有密码子,导致多肽链合成速率的改变进而影响蛋白折叠及构象,但其与DEB表型之间的关系还需要进一步研究。
同时,用Clustal X软件对p.G2006S突变位点氨基酸保守性分析结果显示该位点在进化过程中高度保守。PolyPhen等软件对p.G2006S突变预测结果显示此突变很可能为致病突变;Mutationtaster等软件对c.1731_1732insA突变预测结果显示上述突变可能导致DNA剪接位点、氨基酸序列和蛋白质结构的改变,进一步证实了本研究发现的p.G2006S和c.1731_1732insA突变可能是DEB的致病性突变。
COL7A1基因突变在营养不良性大疱性表皮松懈症中被发现致病的首次报道是在1993年,至2015年3月,人类基因突变数据库报告了700余种基因突变[3,14]。然而如此众多的COL7A1基因突变点所导致的生物学后果也极其复杂[15-17],即基因型-临床表型关系的研究仍处于初始阶段[18-19]。本研究发现了4个尚未见报道过的COL7A1基因突变位点,进一步丰富了COL7A1基因突变数据库,更可为将来开展产前诊断、基因治疗打下基础。
[1] 张希琳,毕新岭,顾军,等.母子二人患不同型营养不良型大疱性表皮松解症[J].临床皮肤科杂志, 2013,42(12):761
[2] TOCKNER B, KOCHER T, HAINZL S, et al. Construction and validation of an RNA trans-splicing molecule suitable to repair a large number of COL7A1 mutations [J]. Gene Ther, 2016, 23(11): 775
[3] CARUANA DM, DAWN G, JURY C. A novel glycine substitution mutation in the COL7A1 gene in two Scottish families with dominant dystrophic epidermolysis bullosa presenting with milia on the hands and feet[J]. Clin Exp Dermatol,2016,41(8):921
[4] SIPRASHVILI Z, NGUYEN NT, GORELL ES, et al. Safety and wound outcomes following genetically corrected autologous epidermal grafts in patients with recessive dystrophic epidermolysis bullosa[J]. JAMA, 2016, 316(17): 1808
[5] BORNERT O, KÜHL T, BREMER J, et al. Analysis of the functional consequences of targeted exon deletion in COL7A1 reveals prospects for dystrophic epidermolysis bullosa therapy[J]. Mol Ther, 2016, 24(7): 1302
[6] ZHU KJ, ZHU CY, ZHOU Y, et al. Case report. Novel and recurrent COL7A1 mutations in Chinese patients with dystrophic epidermolysis bullosa pruriginosa[J]. Genet Mol Res, 2014, 13(3): 7587
[7] JIANG W, SUN TT, LEI PC, et al. Genotype-phenotype correlation in Chinese patients with dystrophic epidermolysis bullosa pruriginosa[J]. Acta Derm Venereol, 2012, 92(1): 50
[8] KIRITSI D, GARCIA M, BRANDER R, et al. Mechanisms of natural gene therapy in dystrophic epidermolysis bullosa[J]. J Invest Dermatol, 2014, 134(8): 2097
[9] SHEN J, ZHANG J, WANG Z, et al. Gene diagnosis and prenatal genetic diagnosis of a case of dystrophic epidermolysis bullosa family caused by gonadosomatic mosaicism for the COL7A1 mutation p.Gly2043Arg in the pregnant mother[J].J Eur Acad Dermatol Venereol,2016,30(9):1627
[10]FORTUNA G, POLLIO A, ARIA M, et al. Genotype-oropharyngeal phenotype correlation in Mexican patients with dystrophic epidermolysis bullosa[J]. Int J Oral Maxillofac Surg, 2014, 43(4): 491
[11]彭炳蔚,朱海霞,李小晶,等.儿童单纯疱疹病毒性脑炎的预后影响因素[J].中华实用儿科临床杂志,2014,29(19):1488
[12]王艳,梁静,赵宝丽,等.第二代测序技术诊断新生儿营养不良型大疱表皮松解症1例及其家系分析[J].临床儿科杂志,2014,32(5):446
[13]MALLIPEDDI R,SUGANTHAN PN,PAN QK,et al.Differential evolution algorithm with ensemble of parameters and mutation strategies[J].Applied Soft Computing,2011,11(2):1679
[14]WU N, JIN L, WANG G. A Novel COL7A1 Mutation in a Chinese family with epider molysis bullosa pruriginosa[J]. Clin Lab,2017,63(1):157
[15]TANG ZL, LIN ZM, WANG HJ, et al. Four novel and two recurrent glycine substitution mutations in the COL7A1 gene in Chinese patients with epidermolysis bullosa pruriginosa[J]. Clin Exp Dermatol, 2013, 38(2): 197
[16]RASHIDGHAMAT E, MCGRATH JA. Novel and emerging therapies in the treatment of recessive dystrophic epidermolysis bullosa[J]. Intractable Rare Dis Res, 2017, 6(1): 6
[17]HAYASHI R, NATSUGA K, WATANABE M, et al. Epidermolysis Bullosa acquisita develops in dominant dystrophic epidermolysis bullosa[J]. J Invest Dermatol, 2016, 136(1): 320
[18]束双双,张连云,黄艳梅, 等.常染色体显性遗传 Alport 综合征一家系 COL4 A3及 COL4 A4的基因突变[J].郑州大学学报(医学版),2014,49(5):723
[19]刘平,卢宁,苏慧, 等.先天性厚甲症2型一家系的基因突变研究[J].西安交通大学学报(医学版),2016,37(1):118
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中华实用诊断与治疗杂志(2022年1期)2022-08-31
临床输血与检验(2022年3期)2022-06-22
山东医药(2021年24期)2021-09-01
诊断学(理论与实践)(2020年1期)2020-04-28
郑州大学学报(医学版)(2019年3期)2019-06-03
中南林业科技大学学报(2019年4期)2019-04-08
国外畜牧学·猪与禽(2018年10期)2018-05-14
森林工程(2018年1期)2018-05-14
国外畜牧学·猪与禽(2018年8期)2018-05-14
