
1例遗传性凝血因子Ⅶ缺陷症患者的家系表型及基因突变分析
2021-09-01 05:30唐兰艳周伟杰罗美玲向利群林发全
山东医药 2021年24期
唐兰艳,周伟杰,罗美玲,向利群,林发全
1广西医科大学第一附属医院检验科,南宁530021;2广西百色市人民医院检验科
凝血因子Ⅶ(FⅦ)是一种维生素K依赖性丝氨酸蛋白酶,由肝脏合成,当血管内皮受损后,组织因子(TF)即被暴露,FⅦ与TF接触后形成TF-FⅦ复合物,后者可直接激活FⅩ和FⅨ,进而生成凝血酶,故FⅦ在外源性凝血途径启动中扮演重要角色。遗传性FⅦ缺陷症是由编码FⅦ基因发生突变而引起的一种罕见出血性疾病[1],呈常染色体隐性遗传,临床出血症状通常分为大出血者、轻微出血者和无症状者。为了预防或治疗遗传性FⅦ缺陷症患者出血及其并发症,主要使用凝血因子替代物来纠正凝血缺陷,包括新鲜冰冻血浆、凝血酶原复合物(PCC)、血浆源性FⅦ浓缩液以及重组活化FⅦ等[1-3]。研究发现,遗传性FⅦ缺陷症患者的出血症状与FⅦ凝血活性(FⅦ:C)存在异质性,单一杂合子多为无症状者,而纯合子与复合杂合子的出血症状常较为严重[4]。2018年5月—2019年7月,本研究对1例遗传性FⅦ缺陷症家系进行实验室表型及基因突变检测,发现先证者及其父母FⅦ:C均降低,尤以先证者FⅦ:C降低最为显著,先证者妹妹FⅦ:C正常;先证者存在FⅦ基因第8外显子p.Thr241Asn杂合错义突变与FⅦ基因第7内含子c.681+1G>T杂合剪接位点突变,先证者父亲携带p.Thr241Asn杂合错义突变,先证者母亲携带c.681+1G>T杂合剪接位点突变,先证者妹妹为野生型,通过分析提示上述突变与先证者及其父母FⅦ:C降低有关。本研究为临床医师评估该类患者在手术期或手术后的出血风险,提前做好预防性治疗,规避出血事件发生,提供了一些数据支持与参考。
1 资料与方法
1.1 临床资料 先证者,男性,9岁,因行包皮环切术就诊于我院泌尿外科。术前凝血检查发现其凝血酶原时间(PT)明显增高,且能被正常混合血浆完全纠正;国际标准化比值(INR)升高;凝血酶原活动度(PTA)显著降低;活化部分凝血活酶时间(APTT)、凝血酶时间(TT)及纤维蛋白原(FIB)均正常。凝血因子检测发现其FⅦ:C显著降低,FⅦ抗原含量(FⅦ:Ag)及其他外源性凝血因子(Ⅱ、Ⅴ、Ⅹ)活性均正常。先证者无自发性皮肤黏膜出血或外伤后出血不止,无肝脏疾病、肺结核和肾炎等病史,未使用抗凝药物,肝功能、肾功能检查结果均正常。术前24 h对先证者予输注600 IU的PCC进行预防性治疗(200 IU/8 h),检测结果显示PT为39.80 s,INR为3.32,PTA为19%。手术过程顺利,出血量约30 mL,无需输血。术后继续输注800 IU的PCC(200 IU/8 h),检测结果显示PT为27.80 s,INR为2.24,PTA为21%,先证者无继发出血,恢复良好。先证者父母为非近亲结婚,先证者有一妹妹,家系成员均无出血及血栓相关疾病史。先证者及其家系成员均已签署知情同意书。
1.2 家系表型分析方法 采集先证者及其家系成员的枸橼酸钠抗凝血各1管,每管2 mL,在IL-ALC TOP700血凝分析仪(美国IL公司)上采用凝固法分别检测PT、INR、PTA、APTT、TT、FIB、因子Ⅱ活性(FⅡ:C)、因子Ⅴ活性(FⅤ:C)、因子Ⅶ活性和因子Ⅹ活性(FⅩ:C);采集先证者及其家系成员、20名健康人的枸橼酸钠抗凝血各1管,每管2 mL,根据《罕见遗传性出血性疾病诊断与治疗中国专家共识(2021年版)》[5],PT纠正试验采用患者血浆与20人份健康人混合血浆按照1:1混合,即刻检测该混合血浆PT,然后置于37℃水浴1、2 h分别检测PT。采集先证者及其家系成员的不抗凝全血各1管,每管5 mL,使用酶联免疫吸附法(中国江苏晶美公司)检测FⅦ:Ag;采集先证者及其家系成员的不抗凝全血各1管,每管5 mL,在HITACHI 7600-020型生化分析仪(日本日立公司)上采用酶法分别检测肝功能与肾功能。所有试剂均为配套试剂,操作步骤均按照仪器的标准操作规程进行。
1.3 FⅦ基因突变分析方法 采集先证者及其家系成员的EDTA抗凝血各1管,每管2 mL,使用吸附柱法提取人类基因组DNA。在illumina HiSeq2500和ABI 3730 DNA分析仪上分别进行高通量测序及Sanger测序,由北京金准基因科技有限公司完成检测。高通量测序发现FⅦ基因外显子及剪切位点突变后,采用Sanger测序验证突变位点,应用Primer Premier 3.0软件设计引物,FⅦ基因第7外显子及侧翼的正向引物:5´-TCCCTCACAAATCTCTGCATC-3´,反向引物:5´-GTGGGCTTGGGGCTGAC-3´;FⅦ基因第8外显子及侧翼的正向引物:5´-AGAGCAC‑GTCTCGGCTAGAG-3´,反向引物:5´-AGCCCCT‑GAGACACTTGAGA-3´,使用Chromas Lite 2.01软件比对测序结果与NCBI基因库公布的参考序列(NG_009262.1),确定突变位点。应用ClustalX2.1软件(http://www.clustal.org/clustal2/)比对来自NCBI数据库10个物种(包括人类、红原鸡、热带爪蟾、黑猩猩、猕猴、家犬、小家鼠、褐家鼠、牛和斑马鱼)FⅦ基因的氨基酸序列。应用FATHMM(http://fathmm.biocompute.org.uk/)、Mutation Assessor(http://mutationassessor.org/r3/)、PolyPhen-2(http://genetics.bwh.harvard.edu/pph2/)、PROVE‑AN(http://provean.jcvi.org/seq_submit.php)和SIFT(http://provean.jcvi.org/protein_batch_submit.php?species=human)五个生物信息学软件分别预测突变氨基酸是否有害或对FⅦ蛋白质功能有无影响。将NCBI数据库公布的FⅦ蛋白参考序列(NP_000122.1)作为模板,应用SWISS-MODEL(https://www.swissmodel.expasy.org/)构建野生型与突变型FⅦ蛋白模型,并使用PyMOL2.2.0软件(https://pymol.org/2/)分析野生型与突变型FⅦ蛋白构象。
2 结果
2.1 家系表型分析结果 先证者PT显著增高,为49.00 s;INR明显增高,为3.86;PT延长能被健康人混合血浆完全纠正;PTA明显降低,为10%;FⅦ:C显著下降,为0.60%;其余指标均正常。先证者父亲FⅦ:C轻度降低,为42.70%;其余指标均正常。先证者母亲FⅦ:C约降至正常值的一半,为29.70%;其余指标均正常。先证者妹妹各项指标均正常。详见表1。
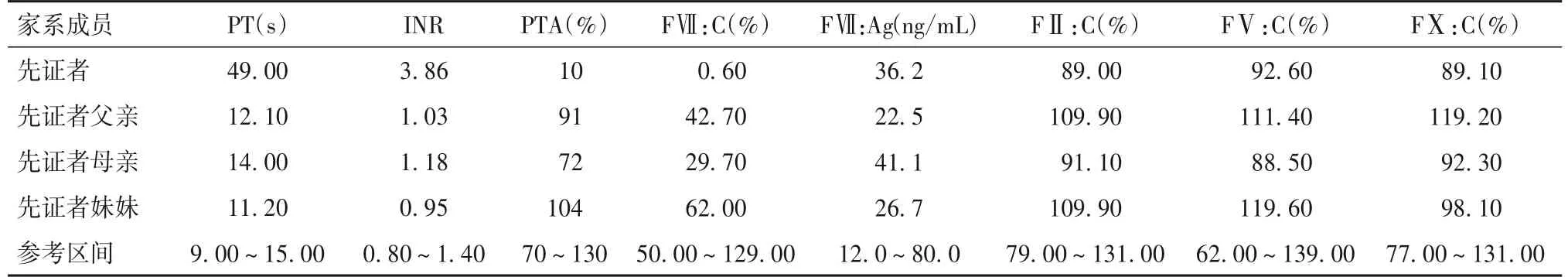
表1 家系部分凝血功能及凝血因子活性结果
2.2 基因突变分析结果 先证者及其父亲FⅦ基因第8外显子均发生c.722C>A杂合错义突变(图1A),即ACC→AAC,导致第241位苏氨酸(Thr)突变为天冬酰胺(Asn);先证者母亲及先证者妹妹为野生型(图1B)。先证者及其母亲FⅦ基因第7内含子均发生c.681+1G>T杂合剪接位点突变(图1C);先证者父亲及先证者妹妹为野生型(图1D)。应用ClustalX2.1软件比对来自NCBI数据库10个物种之间FⅦ基因的氨基酸序列,结果显示Thr241残基保守性较低(图2)。五个生物信息学软件分析结果均提示p.Thr241Asn突变为有害突变或影响蛋白质功能。FⅦ蛋白建模结果显示,发生p.Thr241Asn突变后,Thr241与Val249残基形成的两个氢键和Asn241与Val249残基形成的氢键相似。Thr241残基位于FⅦ蛋白的催化功能区,当发生p.Thr241Asn突变后,Asn241引起Gly177~Pro189肽链出现明显的折叠与空间构象改变。由于c.681+1G>T剪接位点突变会导致10个氨基酸残基(Val218~Gln227)缺失[6],与野生型FⅦ蛋白比较,c.681+1G>T突变型FⅦ蛋白Ser207~Gln227肽链缩短为Ser207~Lys217肽链,导致Cys219与Cys224之间形成的链內二硫键丢失,引起Ser207~Lys217肽链、Gly156~Glu192肽链发生较大的折叠与空间构象改变。与野生型FⅦ蛋白比较,p.Thr241Asn&c.681+1G>T复合杂合突变型FⅦ蛋白Ser207~Lys217肽链(图3A)和Gly156~Glu192肽链(图3B)均发生明显的空间构象变化,且该复合杂合突变导致Gly156~Glu192肽链构象变化与单独c.681+1G>T突变引起该段肽链改变存在一些空间差异。

图1 FⅦ基因第8外显子及第7内含子测序结果

图2 FⅦ基因Thr241残基保守性分析结果
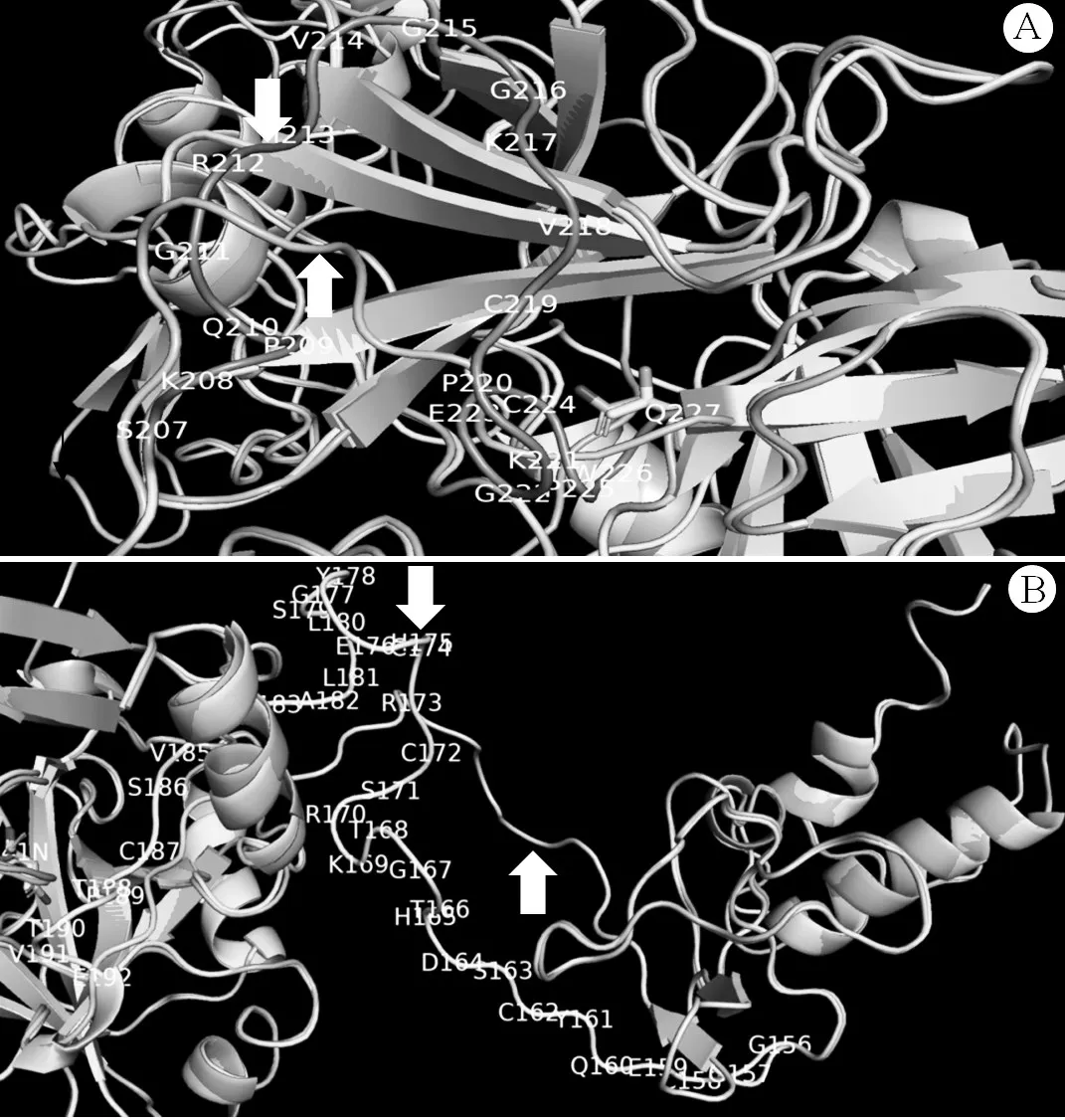
图3 野生型与p.Thr241Asn&c.681+1G>T复合杂合突变型FⅦ蛋白建模结果
3 讨论
FⅦ的编码基因为FⅦ,位于第13号染色体长臂3区4带(13q34),大约距离FⅩ基因上游2.8 kb,由9个外显子和8个内含子组成[7]。FⅦmRNA的选择性剪接可生成含38或60个氨基酸的前导序列,分别由外显子1和2编码,而肝脏中90%的FⅦ转录本不存在外显子2,故前导序列多数情况下为38个氨基酸。外显子3和4编码部分前肽及γ-羧基谷氨酸区,外显子5和6编码两个表皮生长因子样(EGF-1和EGF-2)区,外显子7、8和9编码具有催化作用的丝氨酸蛋白酶区(即催化区),因此成熟的FⅦ由406个氨基酸组成,具有上述4个蛋白结构域[8]。遗传性FⅦ缺陷症由FⅦ突变引起,在罕见遗传性出血性疾病中最为常见,其患病率约为1/500 000[4-5,9],而在一些近亲结婚很频繁的地区或部落,该病患病率可能更高[8,10]。根据FⅦ:C和FⅦ:Ag的质量缺陷不同,遗传性FⅦ缺陷症分为两种类型[11]:Ⅰ型为数量缺陷,即FⅦ凝血活性(FⅦ:C)和FⅦ抗原含量(FⅦ:Ag)两者均降低;Ⅱ型为质量缺陷,即FⅦ:C下降,而FⅦ:Ag正常或接近正常(提示FⅦ蛋白存在功能缺陷)。根据临床表现不同,遗传性FⅦ缺陷症患者分为三类[12]:大出血者,即至少有一种脑出血、胃肠道出血或关节出血的患者;轻微出血者,即无大出血史,但出现月经增多、脐带残端出血、齿龈出血、鼻出血、皮肤黏膜出血或皮肤瘀斑等的患者;无症状者,即证实是遗传性FⅦ缺陷症而无自发轻微或大出血的患者。
遗传性FⅦ缺陷症的出血症状可表现为从无症状到危及生命,严重程度存在明显的个体差异。MARIANI等[13]发现,515例遗传性FⅦ缺陷症患者中39%为无症状者,61%有出血症状,其中常见症状是鼻衄(61%)、皮肤瘀斑(46%)、齿龈出血(30%)、术后出血(25%)和经血过多(在有症状的女性患者中占57.5%)。此外,较常见的出血症状为肌肉血肿、关节出血、血尿、消化道出血和脑出血[5]。通常情况下,FⅦ:C<70%者存在FⅦ缺陷,FⅦ:C<30%者有出血症状[11]。国际血栓止血学会标准委员会根据FⅦ:C水平不同,将遗传性FⅦ缺陷症分为三类[14]:严重缺陷,即FⅦ:C<10%(患者自发性大出血风险最高);中度缺陷,即10%~20%(患者具有轻微自发性或触发性出血风险);轻度缺陷,即20%~50%(患者通常表现为无症状)。尽管如此,FⅦ:C<1%的FⅦ缺陷症患者也可表现为无出血倾向[15],说明FⅦ:C水平与临床出血表型存在异质性,两者之间相关性弱[15-17]。由于再生障碍性贫血、急性髓性或淋巴细胞白血病、骨髓纤维化和造血干细胞移植等患者可发生获得性FⅦ缺陷[18-20],或者患者存在维生素K缺陷、罕见抗凝物等[15],都会造成FⅦ:C水平降低,因此临床医师在诊断遗传性FⅦ缺陷症时,需与上述因素进行鉴别。本研究中先证者为无症状者,PT明显延长,INR明显增高,PTA明显降低,FⅦ:C显著降低,而PT纠正试验、APTT、TT、FIB、FⅦ:Ag、血小板计数和肝功能均正常,与遗传性FⅦ缺陷症的筛选试验结果相符[5]。临床医师综合评估了先证者在围手术期的出血风险,认为其发生出血事件的概率较大,故术前及术后均对先证者予输注PCC进行预防性治疗,手术过程顺利,无需输血,术后也无继发出血,恢复良好。另外,先证者父亲及母亲FⅦ:C均轻度降低,但临床上都无出血症状,其他检测结果均正常,说明血液状态良好,无需特殊处理。
FⅦ突变将导致遗传性FⅦ缺陷症,目前通用突变数据库(UMD)共收录了FⅦ突变800余种[21],其中大部分为错义突变,约占78%,其余为剪切位点突变、小缺失、无义突变等,且以第8外显子发生突变居多[7,17]。本研究中先证者及其父亲FⅦ第8外显子均存在c.722C>A杂合错义突变,导致p.Thr241 Asn,先证者母亲及先证者妹妹均为野生型;还发现先证者及其母亲FⅦ第7内含子均发生c.681+1G>T剪接位点突变,先证者父亲及先证者妹妹均为野生型。根据孟德尔遗传学推测p.Thr241Asn突变来自先证者父亲,c.681+1G>T突变来自先证者母亲。对于p.Thr241Asn突变,国内外已报道三个家系共四例遗传性FⅦ缺陷症患者[4,7,22]。本研究中,同源物种氨基酸多序列比对分析提示Thr241残基保守性不高,我们得知红原鸡、小家鼠、褐家鼠及斑马鱼在第241位置的氨基酸分别为Ser、Val、Val和Val,并非Thr,由此推测单独发生p.Thr241Asn突变对FⅦ蛋白的分泌及稳定性影响较小[7],该推论在先证者及其父亲FⅦ:Ag含量正常中得到了证实。通过五个生物信息学软件分析我们发现,p.Thr241Asn的预测结果较为一致,均显示该错义突变可能为有害突变或影响FⅦ蛋白功能,说明p.Thr241Asn为致病性突变。FⅦ蛋白建模结果显示,Thr241残基位于丝氨酸蛋白酶区(即催化区),处于催化活性中心,发生p.Thr241Asn突变后,由于Thr和Asn均属于极性、中性氨基酸,引起侧链及极性的变化较小,因此野生型Thr241残基、突变型Asn241残基分别与Val249残基形成的氢键相似。将野生型与p.Thr241Asn突变型FⅦ蛋白模型进行比对,我们发现后者Gly177~Pro189肽链发生了明显的折叠以及空间构象的改变,这种改变可能是导致先证者及其父亲FⅦ:C降低的主要原因。c.681+1G>T剪接位点杂合突变首次在一名马来西亚籍遗传性FⅦ缺陷症患者中被报道[7],该患者具有轻度出血症状,FⅦ:C为45%。CAVALLARI等报道了4例无血缘关系的泰国籍遗传性FⅦ缺陷症患者,其中两例是c.681+1G>T纯合子,临床症状分别为胃肠道、颅内出血;第三例是c.681+1G>T与W424X复合杂合子,有颅内出血史;第四例是c.681+1G>T与C389G复合杂合子,症状为轻度出血[6],该研究发现c.681+1G>T激活第7外显子一个隐蔽供体剪接位点,导致一个编码缺失10个氨基酸残基(Val218~Gln227)的FⅦ变异体;还发现c.681+1G>T纯合子虽仅分泌极少量具有功能的FⅦ蛋白,却足以启动凝血,推测该FⅦ变异体与其分泌、活化和催化功能是兼容的。本研究通过FⅦ蛋白建模分析发现,c.681+1G>T突变型和p.Thr241Asn&c.681+1G>T复合杂合突变型FⅦ蛋白Ser207~Gln227肽链均缩短为Ser207~Lys217肽链,造成Cys219与Cys224之间形成的链內二硫键丢失,导致Ser207~Lys217肽链、Gly156~Glu192肽链均发生明显的折叠与空间构象改变,但该两种突变型FⅦ蛋白Gly156~Glu192肽链的空间构象改变存在一些差异,可能是导致先证者与其母亲FⅦ:C水平相差较大的根本原因。
总之,先证者及其父母FⅦ:C均降低,尤以先证者FⅦ:C降低最为显著;先证者发生FⅦ基因第8外显子p.Thr241Asn杂合错义突变与FⅦ基因第7内含子c.681+1G>T杂合剪接位点突变,先证者父亲携带p.Thr241Asn杂合错义突变,先证者母亲携带c.681+1G>T杂合剪接位点突变。
猜你喜欢
中国现代医生(2022年19期)2022-11-04
中国肿瘤临床(2022年14期)2022-08-09
生物化学与生物物理进展(2022年7期)2022-07-25
生物化学与生物物理进展(2022年6期)2022-07-21
临床输血与检验(2022年3期)2022-06-22
昆明医科大学学报(2022年4期)2022-05-23
中国听力语言康复科学杂志(2021年6期)2021-12-21
西南农业学报(2021年10期)2021-12-14
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19
诊断学(理论与实践)(2020年1期)2020-04-28
