
RelA/p65的磷酸化调节及其与肿瘤的关系
2018-06-04 06:26,,*
中南医学科学杂志 2018年2期
,, *
(1.南华大学医学院肿瘤研究所,湖南 衡阳 421001;2.肿瘤细胞与分子病理学湖南省重点实验室)
1 NF-κB概述
核因子-κB(nuclear factor kappa B,NF-κB)是1986年Sen等首先从成熟B淋巴细胞核提取物中检出的一种能与免疫球蛋白κ轻链基因增强子κB序列(GGG ACT TTC)特异性结合并促使κ链基因转录的核蛋白因子,其在几乎所有的动物细胞类型中都被发现,并且涉及对诸如应激、细胞因子、自由基、重金属、紫外线照射、氧化的LDL和细菌或病毒抗原等刺激的细胞反应。近三十年的研究认为它与炎症、免疫反应、创伤、细胞增殖、细胞凋亡以及胚胎发育等重要事件有着密切联系。NF-κB是一种具有多向调节功能的转录因子,主要通过启动和调节多种肿瘤相关基因的转录从而调控细胞的增殖、分化、凋亡及转移。
目前,NF-κB家族共有5个成员:RelA/p65、RelB、c-Rel、p105/p50 (NF-κB1)和p100/p52 (NF-κB2)。RelA/p65、cRel和RelB均在N端含有Rel同源区(Rel homology domain, RHD)和C端的反式激活结构域(transactivation domain, TAD),其中在RHD的C末端有一个核定位区域(nuclear-localization sequence, NLS),负责与DNA结合、二聚体化和核转位,而TAD则与转录活化相关。p50和p52只有RHD而缺乏TAD,因此,p50和p52同源二聚体并不能激活基因转录,而是作为一种抑制分子存在,它们在细胞内通常各自以其前体p105和p100的形式存在。在静息的细胞中,NF-κB 和IκB形成复合体,以无活性形式存在于胞浆中。当细胞受细胞外信号刺激后,NF-κB分别通过经典和非经典途径激活,从而诱导相关基因转录[1]。
研究表明,NF-κB的调节主要是通过多个转录后修饰来实现的。这些修饰可以调控NF-κB信号通路核心成分:IκB激酶复合物(IKK),IκB蛋白和NF-κB亚基的活性。它们包括磷酸化,泛素化,乙酰化,甲基化和亚硝酰化。这些修饰因不同刺激而促成并产生不同的效果,它们彼此并不是孤立的,可通过相互作用形成复杂的网络,共同精细的调节NF-kB的功能。如:RelA/p65的磷酸化就可以调节其乙酰化。磷酸化是这些修饰中一种快速和可逆的酶反应,常作为几种信号转导途径中的关键分子机制。因此,它具有调节转录因子活性的许多优点,并可非常有效的整合来自各种输入信号的信息,而磷酸化过程中单个激酶也可以差异性影响多种转录因子[2]。
以往大多研究都集中在导致NF-κB激活的上游途径[3],由于其在肿瘤发生发展中的促进作用,因此针对抑制NF-κB活性从而用于肿瘤治疗的药物被大量研发。然而,NF-κB抑制剂在肿瘤治疗中的不良疗效及副作用的发现推动着人们对NF-κB更进一步的研究。在很大程度上决定着NF-κB活性并极大的影响其功能的翻译后修饰成为人们研究的热点,而磷酸化是所有修饰中最重要的一种,RelA/p65则是NF-κB最重要的功能亚基,因此,针对RelA/p65的磷酸化开展研究是该领域的热点。
2 RelA/p65的磷酸化调控
2.1RelA/p65NF-κB以二聚体状态存在于细胞中,其中以p65/p50异源二聚体最常见,分布与作用最为广泛,其活性也最强,激活后参与多种基因的表达和调控[1]。p50含有核定位信号,是核因子与DNA结合部位,而p65(RelA)做为最重要的功能亚基[4],不仅含转录活化区域,参与基因转录的起始调节,并可促进p50与DNA结合,且具有特殊的反式激活结构域(TAD),可以调控下游多种靶基因的转录活性,参与细胞多种重要的生命活动,因此成为NF-κB家族研究的焦点,同时也是在NF-κB磷酸化研究中受到最多关注的亚基。一定程度上,可以粗略认为研究RelA/p65的功能即是研究NF-κB的功能。目前,RelA/p65的不同修饰状态研究已被大量报道,对转录活性、蛋白相互作用及降解的不同影响也已被证明。
2.2 RelA/p65各位点的磷酸化近些年对RelA/p65磷酸化大部分研究集中在Ser276和Ser536两个最好理解的磷酸化位点。除此之外,目前所知RelA/p65还有其他11个磷酸化位点。其中Ser205、Thr254、 Ser276 及Ser281四个位点位于RHD,Thr435、 Ser468、 Thr505、Ser529、 Ser535、Ser536及Ser547七个位点则位于TAD区域,而Ser316和Ser311坐落于两者中间靠RHD端的连接处。此外,TAD传统上分为两个不同的反式激活结构域,即TA1和TA2,已被证明是RelA/p65的转录激活区域。TA1位于TAD的C末端部分(残基521-551),TA2位于前90个氨基酸(残基428-521)中[4]。RelA/p65各磷酸化位点见图1。
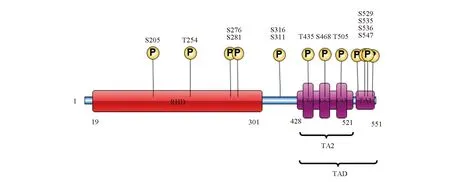
图1 RelA/p65的磷酸化模式图
2.2.1 Ser276 高度保守的Ser276残基位于RHD中,可被LPS、TNF等多种诱导剂刺激,在PKAc作用下磷酸化,从而协调IkB复合物的磷酸化和IKBα的降解[5]。除PKAc激酶外,p65 Ser276磷酸化还由许多其它激酶介导。如:Raf-1、MSK1 和MSK2等激酶。需要说明的是,MSK1/2介导的磷酸化发生在细胞核中。Ser 276的磷酸化在激活RelA/p65和调节其与共激活剂CBP / p300或HDAC共抑制因子的相互作用中起作用。如:Ser276磷酸化触发RelA/p65的构象变化,促进了对NF-κB靶基因启动子的CBP / p300募集,从而增加RelA/p65转录活性[6]。同时,Ser 276的这种磷酸化可被κB-Ras小GTP酶抑制,从而降低与CBP / p300的相互作用,最终导致RelA/p65转录活性抑制[7]。此外,Ser 276磷酸化p65也可以与细胞核中的RelB相关联,该异源二聚体不结合共同的κB位点,从而减轻对TNF的反应活性[4]。Ser276的磷酸化在调节p65的其他翻译后修饰中也是重要的。Ser276磷酸化通过促进与CBP / p300的相互作用而导致p65在K310乙酰化,并可增强转录活性[6]。Ser276磷酸化在调节RelA/p65的转录活性中非常重要,其作为转录激活剂的作用很普遍,当其发生突变时,可致广泛的转录抑制,并可在转基因小鼠中观察到胚胎发育各个阶段的胚胎死亡[8]。Ser276磷酸化还可在PKA激酶作用下促进头颈部鳞状细胞癌恶性表型的形成,表明其对肿瘤的发生发展有促进作用[9]。
2.2.2 Ser-536 Ser536位于RelA/p65反式激活域的TA1亚域,可被IKKα、IKKβ、IKKε、NAK和RSK1等刺激磷酸化,通过增加与CBP / p300的结合和K310乙酰化而增强转录活性,同时对核转位的稳定性有一定影响[5]。在巨噬细胞中,IKKα介导的Ser536磷酸化增加了p65周转,从而降低NF-κB活性并促进炎症因子释放[5]。IKBα的合成所形成的对RelA/p65的负反馈调节是NF-κB调节的重要部分,新合成的IKBα进入细胞核结合NF-κB二聚体并将其转运到细胞质中从而去磷酸化。IκBα的增加可导致转录的减少,然而,IκBα的核积累又可以以基因特异性方式抑制NF-κB活性,导致促炎细胞因子的抑制[10]。这说明,Ser536磷酸化p65与IκBα不相关或不被其调节,Ser536磷酸化可能导致一组不同的靶基因的表达[5]。此外,Ser536磷酸化的p65对IκBα具有较低的亲和力,这导致p65核易位和量的积累增加,更表明IKBα对RelA/p65的负反馈调节是NF-κB调节的重要部分。与Ser276磷酸化可调节乙酰化相似,p65蛋白的糖基化也负调节RelA/p65的Ser536位点磷酸化。Ser536上的糖基化和磷酸化之间存在相互关系,糖基化可抑制其磷酸化,从而抑制后续基因转录[11]。近年研究表明,Ser536磷酸化p65在肿瘤发生发展中既有促进也有抑制作用,已有文献报道其在肝癌中有促进作用[12],而在肠癌、乳腺癌和前列腺癌中则为抑制作用[13],然而瑞典科学家在肠癌中关于Ser536磷酸化的研究呈现相反的结果[14]。以上所述表明Ser536磷酸化p65在肿瘤中的作用不仅与肿瘤类型有关,还可能与患者群体差异也有关。
2.2.3 Thr-505 Thr505位于RelA/p65反式激活域的TA2亚域,可由CHK1激酶介导的ARF诱导而磷酸化,通过增加与HDAC1的相关性而抑制RelA/p65反式激活,其他诱导剂不能激活其磷酸化,可能表明靶向CHK1诱导激活的机制仅存在某些情况下。当DNA损伤时,Thr505的磷酸化可导致NF-kB靶基因的转录抑制(如BcL-xL),并诱导促凋亡基因(如NOXA)。此外,Thr505的磷酸化也负调节自噬、增殖和细胞迁移,表明Thr505磷酸化对于p65的肿瘤抑制活性是重要的[15]。国外文献中p65 Thr505突变小鼠显示的肝损伤时肝细胞增殖和化学诱导肝细胞癌中突变体敏感性增加表明Thr505磷酸化的促凋亡、抑制肝细胞增殖的作用[16]。此外,Thr505磷酸化也可导致HDAC(基因表达阻遏物)增加从而抑制相关基因的表达[4]。
2.2.4 其他磷酸化位点 除了以上位点,其他各位点研究较少。其中Ser468位于RelA/p65反式激活域TAD的TA2亚域的CR2内,可被TNF、IL-1b和T细胞的刺激诱导而磷酸化,它既可以刺激转录激活,也可抑制转录激活。如:TNF-α或IL-1β诱导的IKKβ依赖性磷酸化负面影响p65介导的反式激活,而IKKε触发的T细胞共刺激的磷酸化显示对p65活性的积极影响[16]。Thr435位于p65 TAD的TA2亚结构域CR1内,通过TNFα刺激诱导,可抑制RelA/p65转录激活。Thr435磷酸化的p65降低了与HDAC1的相互作用,从而增强了基因表达[17]。Ser316和Ser311 均位于RelA/p65 同源结构域和转录结构域两者中间靠近RHD的连接点。其中Ser311可被PKC激酶介导的TNF诱导磷酸化,是T细胞中NF-κB活性的重要调节因子[18]。而Ser316的激酶目前未明确,体外分析显示酪氨酸激酶I(CKI)可能作为用IL-1β刺激后Ser316的的激酶,其可在IL-1β的诱导下磷酸化导致转录激活和细胞生长[19]。Ser316可以独立地或与其它磷酸化位点(Ser529和Ser536)协同作用,揭示其有助于不同亚基的NF-κB依赖性基因的表达[19]。Thr254通过TNFα诱导磷酸化,导致与IκBα的结合降低,从而p65核积累,导致p65的稳定性提高和转录增强[11]。Ser529在CK2激酶介导下由IL-1β或TNF-α刺激诱导转录。在U937淋巴细胞中,Ser529被发现在LPS刺激后去磷酸化,而S536则磷酸化增加[5]。Ser547位于RelA/p65反式激活域的TA1亚域中。当DNA损伤,特别是DNA双链断裂时,其可在ATM激酶作用下特异性磷酸化从而诱导转录激活,对IκBα磷酸化或p65核易位无影响[20]。
虽然不同的位点磷酸化后功能不一样,但各位点的磷酸化也不是孤立的,它们的磷酸化通过紧密结合转录共激活因子而增强NF-κB转录反应,这可能表明这些位点修饰的协同效应。例如,当Ser276或311被磷酸化时,RelA/p65与CBP/p300的结合被增强。既往研究已表明各位点磷酸化对NF-κB转录活性具有诱导或抑制的调节作用。最近对不同位点磷酸化与肿瘤关系研究逐渐增多,鉴于不同位点磷酸化对肿瘤的促进或抑制作用(表1),对不同磷酸化位点进行调节的试剂已用于癌症研究。
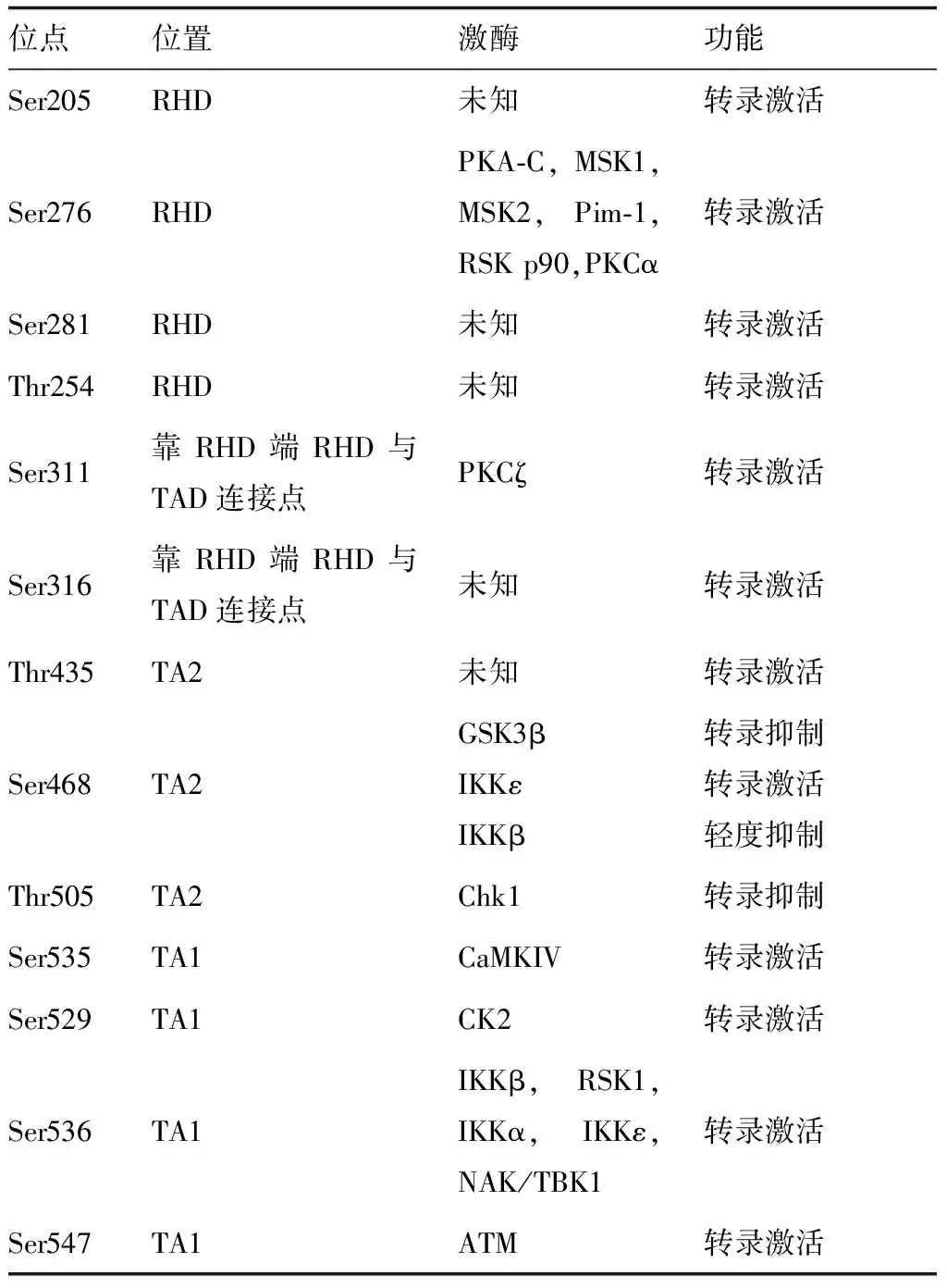
表1 RelA/P65的磷酸化各位点
3 小结与展望
NF-κB从发现迄今已三十余年,由于其抗凋亡和促肿瘤的作用而被当做肿瘤治疗的靶点。早期,大多研究集中在导致NF-κB激活的上游途径,专注于开发高度特异性的对NF-κB活化必须的激酶IKK的抑制剂或阻断IκB降解的抑制剂等(如硼替佐米被成功的用于治疗血液恶性肿瘤)。然而,近年发现这些靶向药物并不是NF-κB转录活性的特异性抑制剂,存在许多脱靶效果,同时也伴随着许多副作用(如:NF-κB负调节IL-1β等细胞因子的分泌,当IKKβ被抑制时,IL-1β等表达水平升高,从而导致许多并发症),这表明单纯的NF-κB抑制治疗是不理想的,需要人们对NF-κB的复杂性有更多的了解。深入研究发现,除肿瘤分期分型、抑癌因子的状态及IKKs存在多种功能等对抗肿瘤药物疗效影响外,NF-κB高效调节及效应与翻译后修饰、其自身状态所处环境、其他信号通路等也有关。其中,NF-κB翻译后修饰一定程度上决定了其转录活性,而磷酸化修饰则是其中最重要的修饰方式。p65/p50作为NF-kB中最广泛、活性最强的异源二聚体,仅p65有转录活性调控域,因此,研究p65很大程度上即研究NF-kB。目前发现RelA/p65有13个磷酸化位点,这些磷酸化位点对RelA/p65的调节起重要作用,调控NF-κB的功能,从而影响下游靶基因的表达。如上所述,在这些已知的位点中:Ser276磷酸化促进头颈部鳞癌的恶性表型;Thr-505磷酸化可对肝癌有抑制作用;而Ser536磷酸化则既可促进也可抑制肿瘤的发生发展。这种不同位点磷酸化有不同作用且就是同一个位点磷酸化在不同癌症作用也不一的发现也证实了NF-kB信号通路的复杂性。NF-κB亚基的位点特异性磷酸化控制与其他因素的相互作用,并对NF-κB二聚体的稳定性、降解和转录活性有影响,而不是作为一个简单的活性开关。因此,在以NF-κB作为治疗靶点时不可单纯的抑制或促进,而应根据磷酸化状态精确考虑。临床上NF-κB抑制剂用于抗肿瘤治疗的疗效不良也证实了这点。对这些基因特异性调节修饰途径的详细了解不仅可以揭示NF-κB的功能与机制,同时也可用于NF-κB相关性疾病的分子分型,进而加速人们对依据肿瘤特定遗传图谱而靶向精准治疗的实践。
参考文献:
[1] ZHANG Q, LENAIDO MJ, BALTIMORE D. 30 Years of NF-κB: a blossoming of relevance to human pathobiology[J]. Cell, 2017, 168(1-2):37.
[2] 李忠, 张培华. 小檗碱通过Toll样受体4/核因子κB信号通路对小鼠病毒性心肌炎发挥保护作用[J]. 中国动脉硬化杂志, 2017, 25(3):250-3.
[3] 赵战芝, 何钒, 唐雅玲,等. PF4通过NF-κB上调巨噬细胞MMP-9表达[J]. 中南医学科学杂志, 2015,43(1):9-13.
[4] PERKINS ND. The diverse and complex roles of NF-kappa B subunits in cancer[J]. Nat Rev Cancer,2012, 12(2):121-32.
[5] FRANK C, SMITH EL, CARMODY RJ. The regulation of NF-κB subunits by phosphorylation[J]. Cells, 2016, 5(1):12.
[6] NIHIRA K, Ando Y, YAMAGUCHI T, et al. Pim-1 controls NF-κB signalling by stabilizing RelA/p65[J]. Cell Death Differ, 2010, 17(4):689-98.
[7] TAGO K, FUNAKOSHI-TAGO M, SAKINAWA M, et al. KappaB-Ras is a nuclear-cytoplasmic small GTPase that inhibits NF-kappaB activation through the suppression of transcriptional activation of p65/RelA[J]. J Biol Chem, 2010, 285(40):30622-33.
[8] DONG J, JIMI E, ZHONG H, et al. Repression of gene expression by unphosphorylated NF-kappaB p65 through epigenetic mechanisms[J]. Genes Dev, 2008, 22(9):1159-73.
[9] ARUN P, BROWN MS, EHSANIAN R, et al. Nuclear NF-kappaB p65 phosphorylation at serine 276 by protein kinase a contributes to the malignant phenotype of head and neck cancer[J]. Clin Cancer Res, 2009, 15(19):5974-84.
[10] GHOSH CC, RAMASWAMI S, JUVEKAR A, et al. Gene-specific repression of proinflammatory cytokines in stimulated human macrophages by nuclear IκBα[J]. J Immunol, 2010, 185(6):3685.
[11] XING D, GONG K, FENG W, et al. O -GlcNAc modification of NF-κB p65 inhibits TNF-α-induced inflammatory mediator expression in rat aortic smooth muscle cells[J]. Plos One, 2011, 6(8):e24021.
[12] SHU G, LANG Z, JIANG S, et al. Isoliensinine induces dephosphorylation of NF-κB p65 subunit at Ser536 via a PP2A-dependent mechanism in hepatocellular carcinoma cells: roles of impairing PP2A/I2PP2A interaction[J]. Oncotarget, 2016, 7(26):40285-96.
[13] BU Y, LI X, HE Y, et al. A phosphomimetic mutant of RelA/p65 at Ser536 induces apoptosis and senescence: an implication for tumor-suppressive role of Ser536 phosphorylation[J]. Int J Cancer, 2016, 138(5):1186-98.
[14] LEWANDER A, GAO J, CARSTENSEN J, et al. NF-κB p65 phosphorylated at serine-536 is an independent prognostic factor in Swedish colorectal cancer patients[J]. Int J Colorectal Dis, 2012, 27(4):447-52.
[15] MSAKI A, SNCHEZ AM, KOH LF, et al. The role of RelA (p65) threonine 505 phosphorylation in the regulation of cell growth, survival, and migration.[J]. Mol Biol Cell, 2011, 22(17):3032-40.
[16] MOLES A, BUTTERWORTH JA, SANCHEZ A, et al. A RelA(p65) Thr505 phospho-site mutation reveals an important mechanism regulating NF-κB-dependent liver regeneration and cancer[J]. Oncogene, 2016, 35(35):4623-32.
[17] O’SHEA JM, PERKINS ND. Thr435phosphorylation regulates RelA (p65) NF-κB subunit transactivation[J]. Biochem J, 2010, 426(3):345-54.
[18] CLAVIJO PE, FRAUWIRTH KA. Anergic CD8+ T lymphocytes have impaired NF-κB activation with defects in p65 phosphorylation and acetylation[J]. J Immunol, 2012, 188(3):1213.
[19] WANG B, WEI H, PRABHU L, et al. Role of novel Serine 316 phosphorylation of the p65 subunit of NF-κB in differential gene regulation[J]. J Biol Chem, 2015, 290(33):20336-47.
[20] SABATEL H, DI VE, GLOIRE G, et al. Phosphorylation of p65(RelA) on Ser(547) by ATM represses NF-κB-dependent transcription of specific genes after genotoxic stress[J]. Plos One, 2012, 7(6): e38246.
(本文编辑:蒋湘莲)
猜你喜欢
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
天津医科大学学报(2021年4期)2021-08-21
天津医科大学学报(2021年3期)2021-07-21
现代仪器与医疗(2021年2期)2021-07-21
世界科学技术-中医药现代化(2021年12期)2021-04-19
昆明医科大学学报(2021年1期)2021-02-07
湖北民族大学学报(自然科学版)(2021年2期)2021-01-15
心肺血管病杂志(2020年5期)2021-01-14
分析化学(2017年12期)2017-12-25
