
单锰取代的Keggin型多酸吸附大气小分子X(X=H2O,N2,O2,NO,N2O,CO 和 CO2)的密度泛函理论计算研究
2018-06-06 05:50刘春光张含玉蒋梦绪
无机化学学报 2018年6期
刘春光 张含玉 蒋梦绪
(东北电力大学化学工程学院,吉林 132012)
0 Introduction
Polyoxometalates(POMs)are early transition metal oxo clusters,which has been widely studied as acid,redox,and bifunctional catalysts in homogeneous as well as in heterogeneous systems[1-6].An advantage of POM-based catalyst is that they can be designed at atomic or molecular levels based on acidic and redox properties.Many physicochemical behaviors of POMs are now successfully explained on a molecular level because of a careful analysis of both experimental and theoretical data[7-11].Keggin-type POMs,incorporating one or several transition metals are subjects of active experimental and theoretical research because they serve as structure models for the active sites of oxygenation or acidic catalysts[12-17].Electronic structure information of these transition-metal-substituted Keggin-type POMs,especially,spin state of the transition metal center,is determining factors for understanding how these POMs perform their catalytic behavior.To date,the accurate determination ofthe relative energies for the different spin states in the different oxidation states of transition metal center in POM species is still a computational challenge because the theoretical treatment of POMs must take into account several intrinsic difficulties[18-19]. POMs are highly negative charge anionic species,involving multiple transition metal atoms.The highly charge POM anions do not exist in gas phase,and only exist in condensed state because the external field generated from the solvent molecules or the counterions stabilizes POM anions.Most of the theoretical investigations for POMs have been done employing conventional density functional theory (DFT)methods with various XC functional,such as,BP86[20],B3LYP[21],BLYP[13],PBE[22],and M06L[14],etc.And the stabilizing fields generated by solvent molecules and counterions were modeled by using continuum model or a set of point charges(modeling the crystal field)[18].The accuracy and deficiency of these methods are reviewed by Poblet and co-workers[18-19].
Mono-transition-metal-substituted Keggin-type POMs have long been referred to as “inorganic metalloporphyrin”because they have same catalytic behaviors in oxygenation catalysis relevant to metalloporphyrins[23-24].In particular,a manganese-substituted Keggin-type POMs showed good activity and high selectivity for the epoxidation of alkenes that compared well to the activity of a manganeseバporphyrin[24].In the past half century,the chemistry of metalloporphyrins has been well studied.It has been found that metalloporphyrins showed highly activity toward oxygen,nitric oxide,carbon monoxide,etc[25-28].Thus,metalloporphyrins have been considered as potential sensing of gases and catalysts for many important reactions,involving reduction ofcarbon dioxide,nitrogen oxides,and the oxidation of hydrocarbons and alcohols,etc[29-34].Compared with metalloporphyrins,the inorganic metalloporphyrins with high redox stability can act as a potential multielectron acceptor.Moreover,the unique withdrawing properties of POM ligands would possibly modify the reactivity of the transition metal center.However,the chemistry of“inorganic metalloporphyrin”,mono-transition-metalsubstituted Keggin-type POMs,is less developed.Undertheguidanceofporphyrin chemistry and motivated by a theoretical work of abdurahman and co-workers[35],a systematic investigation for the ground state electronic structure,relative spin state energies,and adsorption energies of mono-manganese-substituted Keggin-typePOMswith atmosphericsmall molecules X(X=H2O,N2,N2O,CO and CO2)has been performed based on DFT calculations.These findings would provide a basis of understanding the structureproperty relationships for these“inorganic metalloporphyrin”,and be useful to explore the potential applications of POMs in the field of catalytic oxidation of atmospheric small molecules.For modeling of external fields generated by solvent molecules or counterions,the self-consistent reaction field(SCRF)method and a full treatment of the cesium salts Cs4[PW11O39MnⅢX]have both been employed in this work.
1 Computational details
All geometric optimizations were performed by using DFT method with M06L functional.We chose M06L functional[36]because of the proper treatment of transition metal elements and accurate description of non-covalent interactions.Geometric optimization and frequency calculations were used 6-31G(d)basis sets for all main group elements and scalar relativistic effective core potential of LANL2DZ[37-39]for metal atoms.The frequency calculations show that there is no imaginary for all the structures discussed in the present work,which indicates that all of them are minima on the corresponding potential energy surfaces.The adsorption energy(Ead)of the series of atmospheric small molecules X(X=H2O,N2,O2,NO,N2O,CO and CO2)over the porphyrin-like POM ligand was calculated by
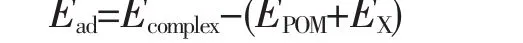
where Ecomplex,EPOM,and EXare the total energies of the metal-X POM complex,POM fragment,and atmospheric small molecules X,respectively.Bulk solvent effects of acetonitrile media have been taken into account via the SCRF method,using the integral equation formalism polarizable continuum model(IEFPCM)solvent model[40]. All calculations were implemented with the Gaussian 09 package[41].
2 Results and discussion
Due to the poly-anionic nature,counterions are always involved in determination of the structure,reactivity,and physicochemical behavior of POMs both in solid and solution[42-44].In the present paper,the interaction between Cs counterions and the surface ofmono-manganese-substituted Keggin-type POMs may form various structures.However,for large-sized POMs,it is difficult to calculate all possible structures because of the high computational cost.Thus,three typical structural patterns have been considered.A aquamanganese derivative Cs4[PW11O39MnⅢH2O]has been firstly employed as an example to probe effects of Cs counterions.As shown in Fig.1,designed around the surface of POM anion,the average distances among the four Cs counterions are becoming more distant in an order of(a)<(b)<(c).According to our DFT-M06L calculations,the distance of the Cs atom and the nearest Obatoms of POM anion is in a range of 0.30~0.31 nm (Obcorresponds to oxygen atom bridging the two tungsten atoms of the POM unit),which is well in agreement with the experimental values in most typical POM cesium salts[45-47].
The water molecule has the singlet ground state(S=0),and is attached to the POM unit[PW11O39MnⅢ]4-as shown in Fig.1.Thus,the 3d electrons of MnⅢcenter have been arranged into three possible spin states,namely,the low-spin state (S=0,MS=1),intermediatespin state(S=1,MS=3),and high-spin state(S=2,MS=5).The spin multiplicities (MS=2S+1),key structural parameters, and calculated relative spin-state electronic energies (Eelec)and total energies involving electronic and zero-point correction energies (Eelec+zero),frontiermolecularorbital (HOMO and LUMO)energies,HOMO-LUMO energy gap,and spin density of the MnⅢcenter(q(Mn))with M06L functional for the three typical structure of cesium salt Cs4[PW11O39MnⅢH2O]in different spin states are reported in Table 1.For the three structures of cesium salt,all parameters listed in Table 1 are almost not sensitive to the change of arrangement of four Cs counterions according to our DFT-M06L calculations.Both Eelecand Eelec+zeroresults indicate that the lowest energy spin state is the high-spin quintet state,and the relative spin-state energy order predicted for those spin states is(S=0)>(S=1)>(S=2)in the three structures of the cesium salt.Meanwhile,a detailed comparison shows that zeroenergy correction leads to a slight increase for the relative spin-state energies.Geometrically,in the three structures of cesium salt,the high-spin ground state has the largest Mn-ligand distance,and the order of the Mn-ligand distances for those spin states is(S=2)>(S=1)≈(S=0).By contrast,the average bond length of 4 Mn-Obbonds (dMn-Ob)is nearly not affected by the change in the spin state.The order of the HOMOLUMO gap for those spin states is(S=1)>(S=2)>(S=0).Mulliken spin density calculations for the open-shell species(triplet and quintet states)indicate that unpaired electrons in both spin states are mainly localized on the MnⅢcenter in the three structures.No large spin contamination for those spin states was found according to our DFT-M06L calculations.
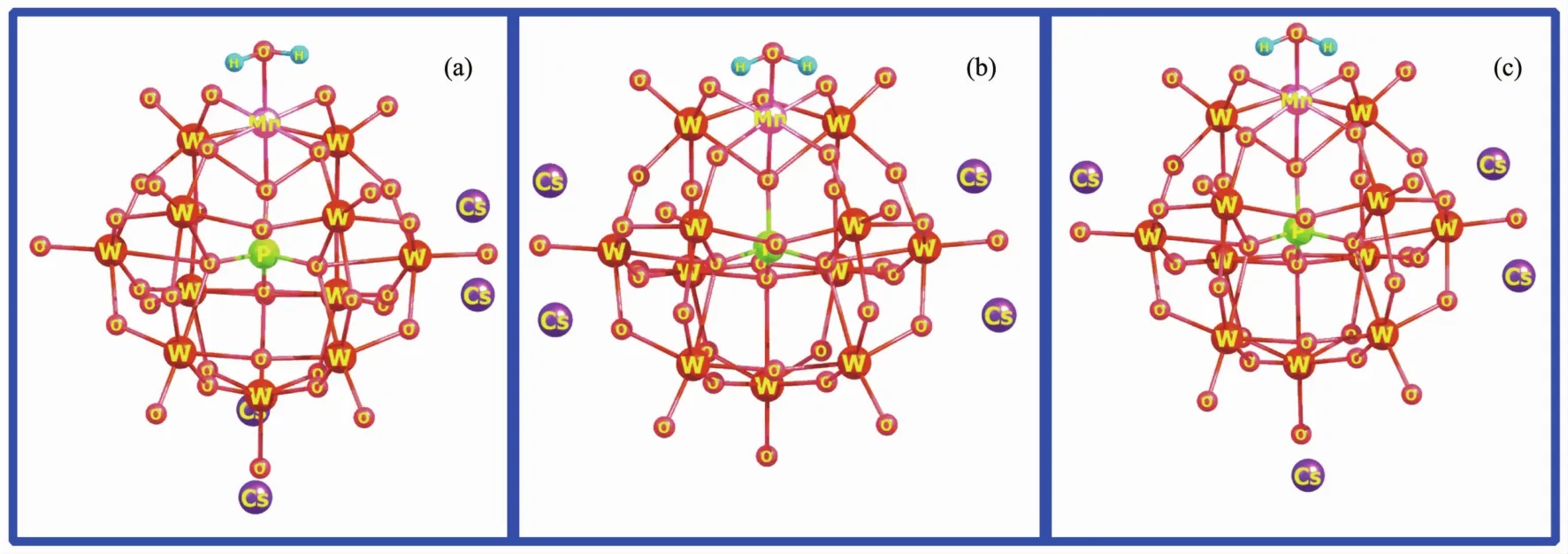
Fig.1 Optimized structures of cesium salt Cs4[PW11O39MnⅢH2O]obtained by M06L/6-31G(d)calculations(LANL2DZ basis sets on metal atoms)
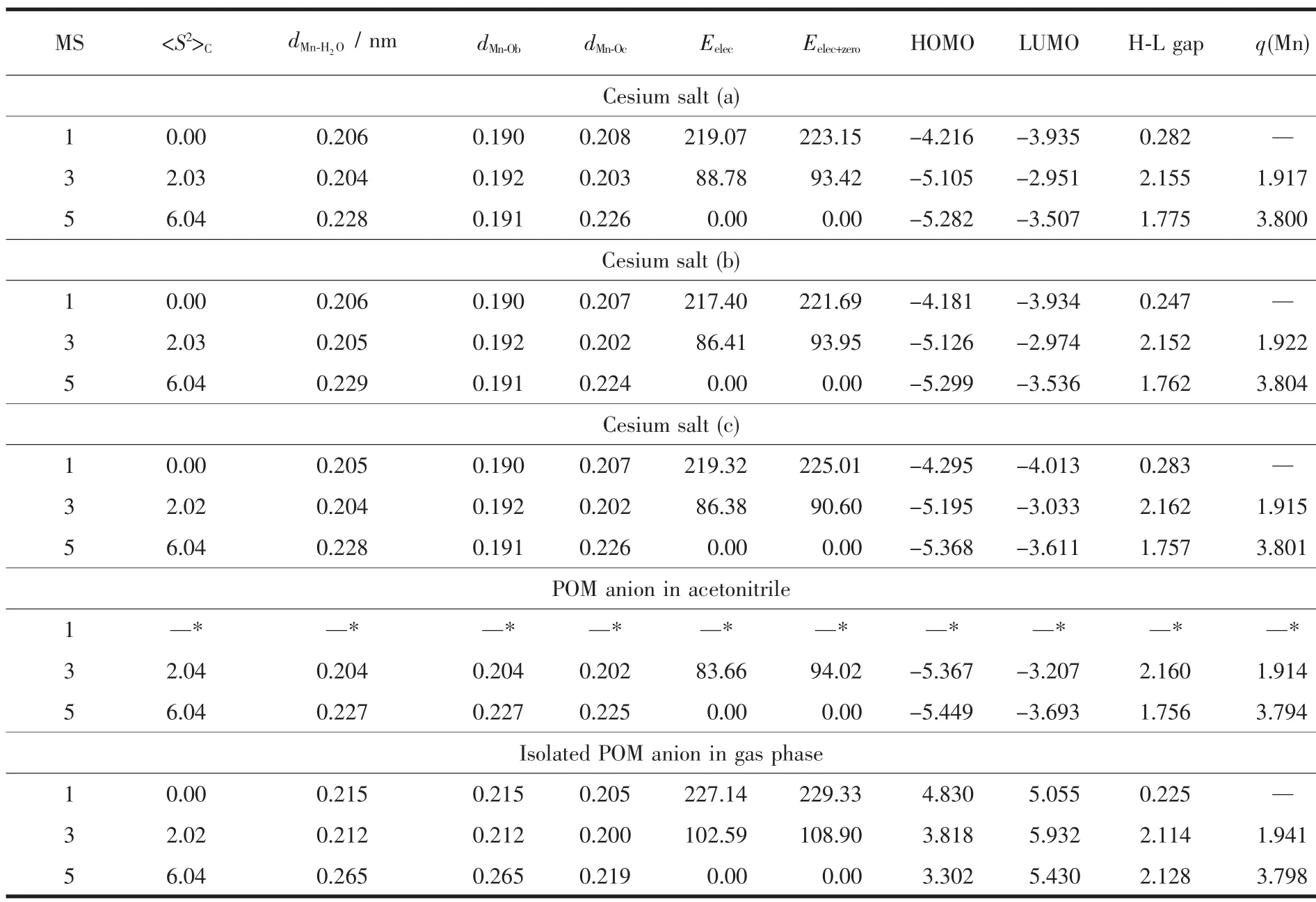
Table 1 Relative energies (kJ·mol-1),key bond length (nm),frontier molecular orbital energy (eV),and Mulliken spin density (a.u.)for three typical structures of cesium salt,isolated POM anion in acetonitrile solution and gas phase
The geometry of POM anion [PW11O39MnⅢH2O]4-also has been optimized both in gas phase and acetonitrile at the same levels,respectively.The gasphase and solventresults forthe POM anion[PW11O39MnⅢH2O]4-are also listed in Table 1.It is worth noting thatgas-phase calculations foran isolated POM anion is the most popular method that has been used on large POM system at an affordable computational cost in the past two decades.Compared with the full treatment of the cesium salt,there was no significant difference for those parameters except for the FMO energies.It has been demonstrated that the instability of isolated POM anions in gas phase is reflected in the high energy of its molecule orbitals[18].As shown in Table 1,our DFT-M06L calculations also reproduce this trend.The calculated HOMO and LUMO levels in gas phase lie at positive energies for those spin states.The stabilizing field generated by solvent molecules and counterions significantly decreases the molecular orbital energies, the calculated HOMO and LUMO for these spin states appearatquite negative energiesin the three structures of cesium salts and solution.But the relative energies of both orbitals(HOMO-LUMO gaps)in these states do not change largely,such as the calculated HOMO-LUMO gap for the triplet state is 2.11,2.16,2.16 eV in gas phase,solution,and cesium salt(b),respectively(Table 1).Due to the low computational cost and reliable electronic and total energy results,the gas-phase calculations will be employed to determine the ground state of the studied POM complexes in the following discussion.Meanwhile,the full treatment of the cesium salt was focused on one structural pattern (c)because those key parameters are almost constant as change of the arrangement of the Cs counterions.
The dinitrogen molecule has a singlet ground state and high chemical inertness.Starting from a linear Mn-N-N arrangement,we performed the geometric optimization of the Mn-dinitrogen POM complex with the three spin states(S=0,MS=1;S=1,MS=3;S=2,MS=5)in gas phase.The predicted lowest energy spin state is the high-spin quintetstate (Supporting Information).The molecular geometry of the studied Mn-dinitrogen POM complex the in its quintet ground state was further optimized in acetonitrile and cesium salt at the same levels,respectively.The calculated key geometric and electronic parameters are listed in Table 2 and Fig.2.As shown in Fig.2,our DFT-M06L calculations show a bent Mn-N-N unit a long Mnligand distance.The calculated Mn-ligand distance decreases in an order of cesium salt(0.30 nm)>gas phase(0.28 nm)>acetonitrile(0.26 nm),indicating a very weak Mn-dinitrogen interaction both in gas phase and condensed state.Mulliken population analysis shows that the spin density of the Mn-dinitrogen POM complex in the quintet ground state is mainly localized on the MnⅢcenter.By contrast,the dinitrogen ligand has very small spin densities.
The ground state of dioxygen molecule is a triplet state with two unpaired elections.In the present paper, we firstly optimized the structure offor the five different spin states (S=0,MS=1;S=1,MS=3;S=2,MS=5;S=3,MS=7;S=4,MS=9)in gas phase to determinate the ground state.As a results,the lowest energy spin state is the triplet state(S=1,MS=3) (Supporting Information).For comparison,the ground state structure was further optimized in acetonitrile and the cesium salt at the same levels,respectively.The calculated key geometric and electronic parameters are also listed in Table 2.It can be found that there are no significant variation of the key geometric and electronic parameters when compared with the gas-phase results.Dioxygen molecule bonding to the Mn center in our studied POM systems leads to a bent Mn-O-O unit with an angle of 117°and a Mn-ligand distance of about 0.20 nm,indicating the dioxygen molecule is loosely bound to the Mn center.Table 2 presents the Mulliken spin densities assigned to the MnⅢcenter and dioxygen ligand in its triplet ground state.For all the studied states,about 3.2 unpaired electrons are localized on the MnⅢcenter,and about 1.5 unpaired electrons with opposite spin are localized on the dioxygen ligand.Thus,the formation of Mn-dioxygen POM complex in itstripletground state may be viewed as an antiferromagnetic coupling between a high-spin MnⅢcenter and a dioxygen molecule.

Table 2 Key bond length (nm),bond angle (°),and Mulliken spin densities (a.u.)for adsorption complexes studied here (C denoted as cesium salt,S denoted as solution,and G denoted as gas phase)
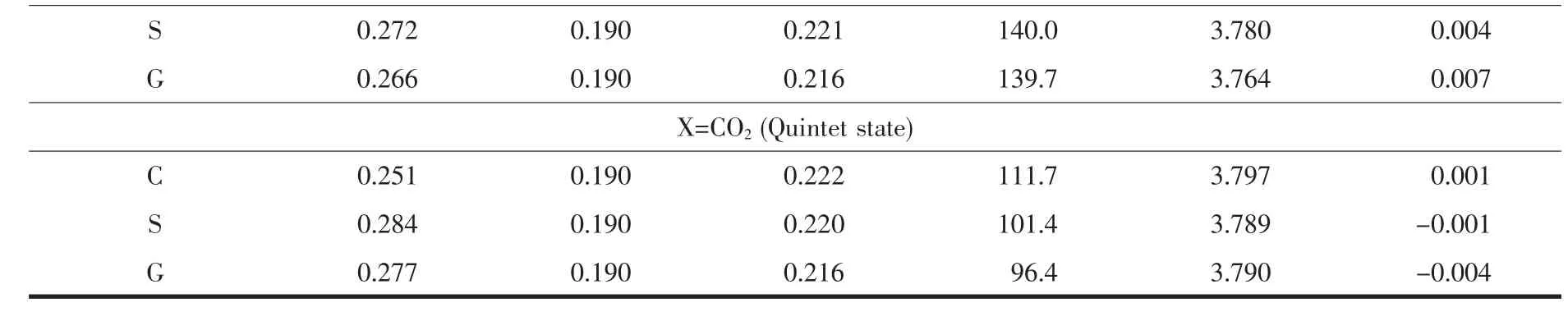
Continued Table 2
NO ligand can be attached to the transition metal center via O-or N-end coordinated models,respectively.In the present paper,we performed the geometric optimization of both coordinated models in the four spin states (S=1/2,MS=2;S=3/2,MS=4;S=5/2,MS=6;S=7/2,MS=8).The lowest spin state is found to be the doublet state with the N-end coordinated model.The ground state molecular geometry is also optimized in two condensed states.The calculated key geometric and electronic parameters are also listed in Table 2.There are no significant variation of the key geometric and electronic parameters in two condensed states when compared with the gas-phase calculations.As shown in Fig.2.The optimized calculations show that coordination of NO ligand to the Mn center leads to a linear Mn-N-O unit with an angle of about 179°and a Mn-ligand distance ofabout0.166 nm,indicating the formation of a strong Mn-N single bond.The Mn-NO moiety in this Mn-NO POM complex may have various electronic structures,such as,MnⅡ-(N≡O)+,MnⅢ-(N=O)·,and MnⅣ-(N=O)-.Table 2 presents the Mulliken spin densities assigned to the MnⅢcenter and NO ligand in its doublet ground state.It can be found that about 1.6 unpaired electrons are localized on the MnⅢcenter,and about 0.8 unpaired electrons with opposite spin are localized on the NO ligand.Thus,coordination of NO ligand to MnⅢcenter in its doublet ground state arises from an antiferromagnetic coupling between an intermediate-spin MnⅢcenter and NO·unit.Therefore,the electronic structure of the Mn-NO moiety of the Mn-NO POM complex in the doublet ground state should be described as MnⅢ-(N=O)·structure.
N2O molecule has a linear structure with a singlet ground state.According to literatures,coordination of N2O molecule to the transition metal center can form metal complexes with O-and N-end models,respectively[48-52].We considered a bent arrangement of Mn-N2O unit as the starting geometry and performed the geometric optimization of both coordinated models in the three spin states (S=0,MS=1;S=1,MS=3;S=2,MS=5)based on gas-phase calculations.The optimized calculations show that the lowest energy spin state is the high-spin quintet state with O-end model.Similarly,the molecular geometry of the Mn-N2O POM complex in its quintet ground state was also optimized in two condensed states.The calculated key geometric and electronic parameters are also listed in Table 2.It can be found that the Mn-ligand distance decreases in an order of acetonitrile(0.27 nm)≈gas phase (0.27 nm)>cesium salt(0.25 nm),which indicates a weak interaction between the N2O ligand and MnⅢcenter both in gas phase and condensed state.Mulliken population analysis also reflects this weak interaction.The spin density of Mn-N2O POM complex in its quintet ground state is mainly localized on the MnIIIcenter,and the N2O ligand has very small spin densities.
Transition metal carbonyl complexes have many types of novel structures because of the variable coordination models of CO ligand.As POM analog,the famous heme-CO systems(an iron porphyrin complex),CO binding to Fe center gives rise to a linear Fe-C-O unit according to many experimental and theoretical studies[53-55].In the present paper,we considered a similar linear structure as the starting geometry for the Mn-CO POM complex in two coordination models(O-and C-end of CO ligand),and performed the geometric optimization in three spin states(S=0,MS=1;S=1,MS=3;S=2,MS=5)based on gas-phase calculations.According to our DFT-M06L calculations,the lowest energy spin state is the high-spin quintet state with the C-end coordination model.Asbefore,theground state molecular geometry of the Mn-CO POM complex was optimized in acetonitrile and the cesium salt at the same levels,respectively.The calculated key geometric and electronic parameters also listed in Table 2 and Fig.2.As shown in Fig.2,our DFT-M06L calculations show a bent Mn-C-O unit with a long Mn-ligand distance.The optimized Mn-ligand distance decreases in an order of acetonitrile(0.27 nm)≈gas phase(0.27 nm)>cesium salt(0.25 nm),indicating a weak interaction between MnⅢcenter and the CO ligand both in gas phase and condensed state.The Mulliken popula-tion analysis also supports this results,where the spin density of the studied Mn-CO POM complex in its quintet ground state is mainly localized on the MnⅢcenter,and the CO ligand has hardly any spin densities.
CO2is a tri-atomic molecule with a linear structure.Due to the different electronegativities,the carbon atom in CO2molecule carries partial positive charge and the oxygen atom in CO2molecule carries partial negative charge.It is known that CO2molecule has two set of π molecular orbitals in its singlet ground state.And both sets of π molecular orbitals are orthogonal to each other.These structural features result in variable coordination models of CO2ligand when interacting with transition metal centers.In the presentpaper,starting from various structures,including linear,bent,O-end,and C-end structures,we performed geometric optimization in three spin states(S=0,MS=1;S=1,MS=3;S=2,MS=5)based on gas-phase calculations. Interestingly, all those optimized calculations provide the same structure as shown in Fig.2.CO2molecule is attached to the MnⅢcenter as a bent arrangement via a very weak interaction in their O-end coordination models,According to our DFT-M06L calculations,the lowest energy spin state is the high-spin quintet state.The molecular geometries of the studied Mn-CO2POM complex in its quintetground state was further optimized in acetonitrile and cesium salt at the same levels,respectively.All the optimized calculations show a long Mn-ligand distance.As shown in Table 2,the optimized Mn-ligand distance decreases in an order acetonitrile(0.28 nm)≈gas phase(0.28 nm)>cesium salt (0.25 nm).Mulliken population analysis also supported this weak interaction.The spin density of the studied Mn-CO2POM complex in its quintet ground state is mainly localized on the MnⅢcenter.There are nearly no spin densities on CO2ligand in its quintet ground state.
On the basis of the optimized geometries in their ground states,the adsorption energy (Ead)of atmospheric small molecules X(X=H2O,N2,O2,NO,N2O,CO,and CO2)over the porphyrin-like POM ligand was calculated in gas phase,acetonitrile,and cesium salt,respectively.The calculated Eadvalues were listed in Table 3.It can be found that the solvent calculations for each POM complex studied here provided a low adsorption energy when compared with the results derived from gas phase and a full treatment with the cesium salts.All DFT-M06L calculations indicate the adsorption energy of the POM complexes studied here increases in the following order:N2<N2O<CO≈CO2<O2<H2O<NO,especially,the Mn-NO POM complex provides considerable adsorption energy of-199.6,-175.5,and-206.5 kJ·mol-1in gas phase,acetonitrile,and cesium salt,respectively.This result is well in agreement with the prediction based on their molecular geometries,a strong Mn-N single bond.

Table 3 Calculated adsorption energies (kJ·mol-1)for the series of POM complexes in gas phase,acetonitrile,and cesium salt obtained by M06L/6-31G(d)calculations (LANL2DZ basis sets for metal atoms)
3 Conclusions
We have performed DFT calculations to study molecular geometries and electronic structures of a series of Mn-POM complexes[PW11O39MnⅢX],which are generated by coordination of atmospheric small molecule X(X=H2O,N2,O2,NO,N2O,CO and CO2)to the MnⅢcenter.Because those Mn-POM complexes carry highly negative charge,we firstly explored the counterions effects by means of a full treatment of the cesium salt Cs4[PW11O39MnⅢH2O].Three typical structure patterns of the cesium salt,which were differentiated by the average distances among the four Cs counterions around the surface of POM anion,were considered in the present work.According to our DFT-M06L calculations,the key geometric and electronic parameters are almost constants as change of the average distances of the four Cs counterions.For comparison,molecular geometry of[PW11O39MnⅢH2O]4-was also optimized in both gas phase and acetonitrile.Optimized calculations show that there are no significant variation of the key geometric and electronic parameters when compared with the full treatment of the cesium salt.DFT-derived relative energy of different spin states reveal that the lowest energy spin state is the high-spin quintet state for(X=H2O,N2,N2O,CO,and CO2),triplet state forand doublet state for[PW11O39MnⅢNO]4-,respectively.Our DFT-M06L calculations indicate that the adsorption energy of those atmospheric small molecules over the porphyrin-like POM ligand increases in the following order:N2<N2O<CO≈CO2<O2<H2O<NO.Mulliken population analysis shows that coordination of NO ligand to MnⅢcenter in its doublet ground state arises from an antiferromagnetic coupling between an intermediate-spin MnⅢcenter and NO·unit.
Supporting information is available at http://www.wjhxxb.cn
[1]Kozhevnikov I V.Chem.Rev.,1998,98:171-198
[2]Weinstock I A.Chem.Rev.,1998,98:113-170
[3]Mizuno N,Misono M.Chem.Rev.,1998,98:199-218
[4]Dolbecq A,Dumas E,Mayer C R,et al.Chem.Rev.,2010,110:6009-6048
[5]Sun M,Zhang J,Putaj P,et al.Chem.Rev.,2014,114:981-1019
[6]Wang S S,Yang G Y.Chem.Rev.,2015,115:4893-4963
[7]Vilà-Nadal L,Mitchell S G,Rodríguez-Fortea A,et al.Phys.Chem.Chem.Phys.,2011,13:20136-20145
[8]Vilà-Nadal L,Mitchell S G,Long D L,et al.Dalton Trans.,2012,41:2264-2271
[9]McGlone T,Vilà-Nadal L,Miras H N,et al.Dalton Trans.,2010,39:11599-11604
[10]Rubinstein A,Jiménez-Lozanao P,Carbó J J,et al.J.Am.Chem.Soc.,2014,136:10941-10948
[11]Marrot J,Pilette M A,Haouas M,et al.J.Am.Chem.Soc.,2012,134:1724-1737
[12]FAN Ying(樊莹),LIU Shi-Zhong(柳士忠).Chinese J.Inorg.Chem.(无机化学学报),2002,18:635-638
[13]Khenkin A M,Kumar D,Shaik S,et al.J.Am.Chem.Soc.,2006,128:15451-15460
[14]Liu C G,Liu S,Zheng T.Inorg.Chem.,2015,54:7929-7935
[15]Liu C G,Su Z M,Guan W.Inorg.Chem.,2009,48:541-548
[16]Liu C G,Guan W,Yan L K,et al.Dalton Trans.,2011,40:2967-2974
[17]Liu C G,Guan W,Yan L K,et al.Dalton Trans.,2009,38:6208-6213
[18]Poblet J M,López X,Bo C,et al.Chem.Soc.Rev.,2003,32:297-308
[19]López X,Carbó J J,Bo C,et al.Chem.Soc.Rev.,2012,41:7537-7571
[20]Maestre J M,López X,Bo C,et al.J.Am.Chem.Soc.,2001,123:3749-3758
[21]Antonova N S,Carbó J J,Kortz U,et al.J.Am.Chem.Soc.,2010,132:7488-7497
[22]Efremenko I,Neumann R.J.Am.Chem.Soc.,2012,134:20669-20680
[23]Hill C L,Brown R B.J.Am.Chem.Soc.,1986,108:536-538
[24]Mansuy D,Bartoli J F,Battioni P,et al.J.Am.Chem.Soc.,1991,113:7222-7226
[25]Karpuschkin T,Kappes M M,Hampe O.Angew.Chem.Int.Ed.,2013,52:10374-10377
[26]Chen O,Groh S,Liechty A,et al.J.Am.Chem.Soc.,1999,121:11910-11911
[27]Kano K,Itoh Y,Kitagishi H,et al.J.Am.Chem.Soc.,2008,130:8006-8015
[28]Wasser I M,Huang H W,Moenne-Loccoz P,et al.J.Am.Chem.Soc.,2005,127:3310-3320
[29]Morris A J,Meyer G J,Fujita E.Acc.Chem.Res.,2009,42:1983-1994
[30]Groves J T,Roman J S.J.Am.Chem.Soc.,1995,117:5594-5595
[31]Saito S,Ohtake H,Umezawa N,et al.Chem.Commun.,2013,49:8979-8981
[32]Phougat N,Vasudevan P,Jha N K,et al.Transition Metal Chem.,2003,28:838-847
[33]Meunier B.Chem.Rev.,1992,92:1411-1456
[34]Liu L,Yu M,Wayland B B,et al.Chem.Commun.,2010,46:6353-6355
[35]Abdurahman A,Renger T.J.Phys.Chem.A,2009,113:9202-9206
[36]Zhao Y,Truhlar D G.J.Chem.Phys.,2006,125:1-18
[37]Hay P J,Wadt W R.J.Chem.Phys.,1985,82:270-283
[38]Wadt W R,Hay P J.J.Chem.Phys.,1985,82:284-293
[39]Hay P J,Wadt W R.J.Chem.Phys.,1985,82:299-310
[40]Tomasi J,Mennucci B,Cammi R.Chem.Rev.,2005,105:2999-3093
[41]Frisch M J,Trucks G W,Schlegel H B,et al.Gaussian,Inc.,Wallingford CT,2009,Gaussian 09,Revision D.01
[42]Kirby J F,Baker L C W.Inorg.Chem.,1998,37:5537-5543
[43]Brevard C,Schimpf R,Tourne G,et al.J.Am.Chem.Soc.,1983,105:7059-7063
[44]Grigoriev V A,Cheng D,Hill C L,et al.J.Am.Chem.Soc.,2001,123:52925307
[45]Kato C N,Kashiwagi T,Unno W,et al.Inorg.Chem.,2014,53:4824-4832
[46]Mizuno N,Min J S,Taguchi A.Chem.Mater.,2004,16:2809-2825
[47]Bi L H,Reicke M,Kortz U,et al.Inorg.Chem.,2004,43:3915-3920
[48]Pamplin C B,Ma E S F,Safari N,et al.J.Am.Chem.Soc.,2001,123:8596-8597
[49]Groves J T,Roman J S.J.Am.Chem.Soc.,1995,117:5594-5595
[50]Groves J T,Quinn R.J.Am.Chem.Soc.,1985,107:5790-5791
[51]Ben-Daniel R,Weiner L,Neumann R.J.Am.Chem.Soc.,2002,124:8788-8789
[52]Ghosh A.Acc.Chem.Res.,2005,38:943-954
[53]Kachalova G S,Pepov A N,Bartunik H D.Science,1999,284:473-476
[54]Spiro T G,Kozlowski P M.J.Am.Chem.Soc.,1998,120:4524-4525
[55]Rovira C,Kunc K,Hutter J,et al.Int.J.Quantum Chem.,1998,69:31-35
猜你喜欢
中国机械工程(2022年22期)2022-11-25
中国机械工程(2022年7期)2022-04-20
当代陕西(2022年5期)2022-04-19
小读者(2021年4期)2021-06-11
考试与评价·高一版(2020年4期)2020-11-12
学生天地(2020年31期)2020-06-01
中国机械工程(2019年17期)2019-09-19
学生天地(2019年30期)2019-08-25
中国机械工程(2018年4期)2018-03-06
37°女人(2015年4期)2015-11-04
