
蛋白芯片技术检测幽门螺杆菌抗体200例结果分析
2018-09-20 01:04贾玉婷徐晓雯
中国实验诊断学 2018年8期
贾玉婷,徐晓雯,余 瑶,王 丹
(吉林大学第一医院 胃肠内科,吉林 长春 130021)
幽门螺杆菌(H.pylori)感染已被公认是引起慢性胃炎、消化性溃疡的主要病因[1,2]。但并非所有患者感染后都会致病,是否致病除了与宿主本身的个体差异及环境等因素有关外,所感染菌株自身的基因型也是一个不可忽略的因素。H.pylori本身含有细胞毒相关基因A(CagA)、空泡毒素基因A(VacA)及尿素酶 (Ure) 等多种抗原蛋白,这些抗原可以刺激机体免疫系统,从而产生针对不同抗原决定簇的相应的多种抗体。研究表明不同的抗体与H.pylori感染所致的疾病严重程度及预后有密切的关系[3],本研究通过测定血清中CagA、VacA及Ure三种抗体水平,探讨不同毒力基因型H.pylori菌株的感染与胃十二指肠疾病严重程度之间的相关性。
1 材料与方法
1.1 病例资料
收集2012年1月至2016年1月就诊于吉林大学第一医院胃肠内科门诊或住院的200例H.pylori感染患者,均经过尿素14C呼气试验检测阳性,且同期胃镜证实为慢性胃炎或消化性溃疡的患者。排除检查前4周内服用过抗菌药物、铋剂、质子泵抑制剂或可能干扰检测结果的中药制剂等,或既往有消化道手术史者。
1.2 研究方法
蛋白芯片系统检测血清H.pylori抗体:严格按照西安联尔生物科技有限公司提供的H.pylori抗体蛋白芯片检测试剂盒说明书操作,用配套的生物芯片阅读系统对结果进行分析。受试者清晨空腹采集静脉血2 ml,4 h内以3 000 r/min的转速离心10 min,分离血清后立即进行蛋白芯片检测。检测步骤如下所述:(1)在芯片的上片盒窗口内滴加4滴试剂A,使膜表面完全浸湿;(2)待完全渗入后,室温放置1 min,加待检血清100 μl;(3)待血清完全渗入后,再滴加6滴试剂B;(4)待试剂B完全渗入后,滴加10滴试剂C;(5)待试剂C完全渗入后,最后滴加6滴试剂D;(6)在反应完毕后30min之内,将芯片放入芯片阅读系统进行分析。通过Biochip system 2.0软件分析患者血清中CagA、VacA、Ure三种抗体感染率。
1.3 统计学分析
使用SPSS 19.0统计软件进行数据分析。计数资料用百分率表示,不同率的比较采用χ2检验;计量资料以mean±SD表示,组间比较采用t检验。P<0.05为差异有统计学意义。
2 结果
2.1 一般情况
采集200例患者,其中男97例,女103例,年龄18-75岁,平均年龄(43.86±13.80)岁。其中十二指肠球部溃疡(duodenal ulcer,DU)41例、胃溃疡(gastric ulcer,GU)8例,复合型溃疡(compound ulcer,CU)12例,慢性胃炎-萎缩性(chronic atrophic gastritis,CAG)20例、慢性胃炎-非萎缩性(chronic non-atrophic gastritis,CNAG)119例。
2.2 不同毒力因子的分布情况
2.2.1CagA抗体分布情况 CagA抗体总阳性率为19%,DU、GU,CU,CAG、CNAG的CagA抗体阳性率分别为31.7%、25%、25%、25%、12.6%,DU、GU、CU、CAG的CagA抗体检出率明显高于CNAG(P<0.001),而DU的检出率更明显;结果见表1。
2.2.2VacA抗体分布情况 VacA抗体总阳性率为39.5%,5组VacA抗体阳性率分别为56.1%、37.5%、50%、40%、32.8%,各组之间的差异无统计学意义(P=0.110),结果见表1。
2.2.3Ure抗体分布情况 Ure抗体总阳性率为75.5%,五组的Ure抗体阳性率分别为80.5%、75%、75%、75%、73.9%,差异无统计学意义(P=0.654)。结果见表1。
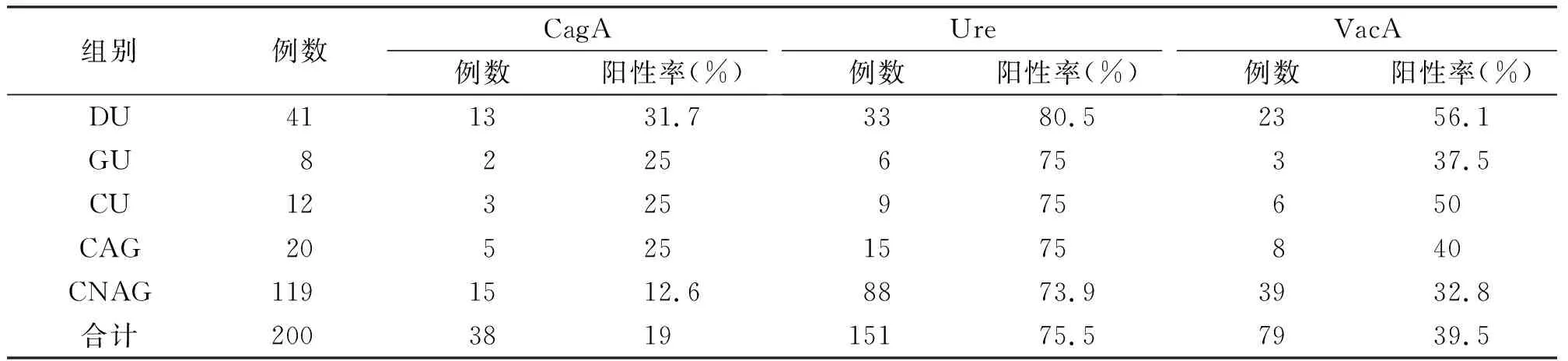
表1 H.pylori阳性的胃十二指肠疾病中毒力因子的分布情况
3 讨论
H.pylori在我国感染率仍高达约50%[4],而目前检测的方法主要有侵入性和非侵入性两种,各有优缺点。侵入性方法主要包括快速尿素酶试验、胃黏膜组织切片染色镜检、细菌培养以及放大染色内镜的应用,这类方法均通过胃镜检查和(或)胃黏膜活检取样,虽然具有较高的准确性,但部分患者因恐惧心理无法耐受,或检查过程中可能出现H.pylori或其它病原菌交叉感染,或缺乏放大或电子染色内镜的相应设备等原因,使得临床常规应用受到了限制。非侵入性的检测方法包括13C或14C尿素呼气试验、粪便抗原试验和血清学试验等。其中蛋白芯片技术是一种集快速、简便、无创、准确、费用低等优点于一体的血清学检测方法,只需采集患者的血清即可,无放射性损伤,而且可以避免交叉感染。以往认为血清学检查只能用于流行病学调查,而不适于临床诊断,但通过大量的研究表明,H.pylori血清学筛查,能够发现所感染H.pylori不同毒力基因型,结合不同基因型与疾病严重程度的相关性,不但可以预测疾病风险指导患者进一步检查及得到相应的根除治疗,同时也对反复根除失败的患者提供继续治疗的风险效益评估。本研究通过蛋白芯片方法快速检测出H.pylori多种抗体,发现感染不同毒力基因型与胃炎或消化性溃疡有一定的相关性,可以作为临床风险效益评估的辅助手段。
H.pylori通过“口-口”或“粪-口”等传播途径进入患者胃内,一部分被胃酸杀灭,另一部分依靠其鞭毛穿过胃黏液层,定值于胃黏膜上皮细胞表面,不但可以避免胃酸对其的杀灭作用,与此同时机体的免疫机能也很难将其根除。由于其自身含有CagA、VacA、Ure等多种抗原蛋白,不同毒力基因型的H.pylori通过各自不同的机制促使胃、十二指肠黏膜发生炎症反应,进而导致了感染后出现不同的临床反应,即基因型的多态性导致了致病的差异性,大量的研究结果[5-10]表明,CagA阳性的H.pylori为高毒力株,与萎缩性胃炎、消化性溃疡的发生及与疾病的发展和愈后有着密切的关系,即Cag A阳性的H.pylori感染者有更明显的消化道粘膜损伤。陈周利[11]等的研究表明,CagA毒力蛋白抑制了人体内IL-9的表达,而IL-9可能对H.pylori感染所致的黏膜损害发挥抑制作用保护胃黏膜,感染后患者血清中IL-9减少,胃黏膜失去了其保护作用,故感染CagA阳性的H.pylori易发生黏膜的损伤。另有研究表明[12]Cag A抗原诱导消化道黏膜中细胞因子IL-8 的高表达,而IL-8可趋化中性粒细胞在消化道黏膜中浸润,通过促进中性粒细胞的聚集和活化启动炎症过程,从而导致了消化道黏膜上皮的损害,出现相应的消化道不适症状。本研究中应用蛋白芯片技术对明确诊断H.pylori感染患者的血清中多种抗体进行检测,其中200例患者中CagA抗体总阳性率为19%,相对偏低,与文献报道不同[6,7],分析原因可能与本地区感染的菌株基因型不同有关,相关研究结果也表明[13],H.pylori感染存在地域性差异,不同地区存在不同基因型的优势菌种,其所含CagA也不尽相同,直接影响检测结果。CagA抗体总阳性率虽然偏低,但在DU、GU、CU、CAG中 CagA抗体阳性率却显著高于CNAG(P<0.001),表明CagA与疾病的严重程度及预后有着密切的关系,在判断疾病的严重程度上仍有一定价值,Cag A抗体阳性的H.pylori感染者有更明显的消化道粘膜损伤,必须予以积极的根除治疗。提示临床上通过CagA的检测对反复根除失败的患者是否继续根除治疗有一定的指导意义。VacA是一种95 kD的蛋白质毒素,其作用主要是引发促分裂原活化蛋白激酶激活,细胞膜通透性增高,细胞内形成空泡,诱导细胞凋亡和免疫功能紊乱导致免疫损伤,并因此协助细菌逃避机体的免疫监视[14,15]。因为VacA基因的多态性,使其在有毒力的H.pylori菌株中作用效能存在着较大的差异。而几乎所有的H.pylori均含有尿素酶,其可分解尿素产氨,导致细菌微环境pH值增高,使细菌能够穿过胃黏液层到达黏膜表面定值和损伤胃黏膜。本研究中VacA、Ure抗体总阳性率为分别为39.5%和75.5%,5组VacA、Ure抗体阳性率之间差异无统计学意义(P=0.110,P=0.654),VacA、Ure在5种胃十二指肠疾病之间未显示明确相关性,提示VacA、Ure阳性的H.pylori感染者,利用其判断慢性胃炎或消化性溃疡的严重程度及愈后方面作用有限。随着临床H.pylori治疗的广泛开展,需要指导后续治疗的反复根除治疗失败者、根除治疗风险增加的老年感染者均需要进行个体获益-风险综合评估,实现个体化处理,而血清学抗体检测的发展和完善,无疑会成为未来重要的辅助评估手段。
综上所述,蛋白芯片技术检测多种抗体并进行毒力分型,对H.pylori感染的诊断、评估预后及指导治疗有着重要的临床意义;不同抗体的存在与各种胃十二指肠疾病的发生关系密切,其中毒力因子CagA抗体阳性的菌株感染与消化性溃疡、慢性萎缩性胃炎的发生密切相关。未来开展H.pylori不同基因分型菌株在早期胃癌中的表达研究将成为新的探索方向。
猜你喜欢
今日畜牧兽医(2022年10期)2022-12-23
传染病信息(2022年4期)2022-11-23
河南医学研究(2022年18期)2022-09-30
健康护理(2022年5期)2022-05-26
中华养生保健(2020年2期)2020-11-16
农药科学与管理(2019年9期)2019-11-23
农药科学与管理(2019年6期)2019-11-23
中国抗生素杂志(2019年6期)2019-07-06
中国防痨杂志(2018年3期)2018-03-07
中国人兽共患病学报(2017年11期)2017-12-13
