
QuEChERS EMR-Lipid结合LC-MS/MS测定鸡蛋中磺胺类和喹诺酮类药物残留
2018-10-13 05:55刘红曾建勇梁雪琪邓晓华李震源
现代食品科技 2018年9期
刘红,曾建勇,梁雪琪,邓晓华,李震源
(1.中山市农业科技推广中心,广东中山 528403)(2.中山市农产品质量监督检验所,广东中山 528400)
磺胺类抗生素(sulfonamides,sAs)是人工合成的具有对氨基苯磺酰胺结构的一类抗菌药物[1],喹诺酮类(4-quinolones)是一类含4-喹诺酮基本结构的抗菌药物[2],两类药物都因具有化学性质稳定、抗菌谱广、高效、低毒、价格低和使用方便等优点而被广泛应用到养殖业上,用来治疗和预防动物的疾病感染,并越来越受到养殖户的重视。但由于生产者用药不规范或不遵守正确的休药期等因素,容易导致 sAs和 FQs(fluoroquinolones,氟喹诺酮类抗生素)在畜禽体内过量积聚残留,污染动物性食品,间接对人体产生危害,如过敏反应、泌尿系统损害等,严重的可导致休克甚至死亡。某些药物还存在潜在的致癌性,对人体产生持久危害。为此,世界各国对食品中的sAs和FQs的残留限量提出了严格要求,而且许多国家将sAs和FQs列入抗生素禁用清单。目前,欧盟、美国、日本和韩国等国家均将磺胺类药物列为动物饲养过程中限制使用的药物,美国则禁止喹诺酮类药物应用于动物源性食品[3],我国农业部《兽药停药期规定》中也指出多种喹诺酮药物制剂在产蛋鸡中禁用,农业部公告第235号对动物源性食品中使用磺胺类药物的规定限量为总量不得大于100 μg/kg。
目前,国内外对磺胺类药物和喹诺酮类药物残留检测研究比较常见,报道过的文献也较多,但多数都是针对其中一类药物的检测分析研究,两类药物一起进行前处理检测的并不多见。国内外对sAs和FQs的分析检测方法主要有微生物检测法[4~6]、酶联免疫吸附法[7,8]、毛细管电泳法(CE)[9,10]、高效液相色谱法(HPLC)[11~20]及高效液相色谱-串联质谱法(HPLC-MS/MS)[21~29]等,其中,HPLC-MS/MS 法是检测动物源性食品中兽药残留的重要方法,这种方法结合液相保留时间,并采用多重反应监测(MRM)模式,对特征离子进行定量和定性分析,适合于复杂基质样品的检测。但目前检测sAs和FQs的LC-MS/MS方法主要集中在水产品、鸡肉、猪肉和牛奶等方面,未有文献报道鸡蛋中磺胺类药物的液相色谱-质谱/质谱法,可以同时检测鸡蛋中磺胺类药物和喹诺酮类药物的快捷方法更未见文献报道。
在样品前处理过程中,本实验采用了创新型安捷伦增强型脂质去除产品(QuEChERS EMR-Lipid)萃取技术净化样品。与传统技术相比,它是一种能够选择性去除复杂基质中脂质并挑战鳄梨等高脂肪含量样品的独特吸附剂,可针对任意样品前处理产品实现最彻底的脂质去除和分析物回收,因此您可以在不损失分析物的前提下去除脂质。此技术不仅提高了方法的灵敏度、精密度和回收率,而且其操作步骤也比较简单,减少了操作过程中的各种误差,样品前处理的时间也大大缩短。
鉴于此,本文旨在建立一种新的高效液相色谱-三重四级杆串联质谱联用法以检测禽蛋中多种磺胺类药物和多种氟喹诺酮类药物的残留含量,同时结合最新的 QuEChERS EMR-Lipid快速样品提取技术进行样品前处理。此方法不仅在结果上大大提高了药物的检测灵敏度,在样品前处理上也适应了快速、简便、准确度高的优点,适应了食品安全检测技术简便快捷和自动化的发展趋势。
1 材料与方法
1.1 材料与试剂
PRIME-HLB 固相萃取柱(60 mg/3 cc),购自Waters公司;QuEChERS EMR-Lipid快速萃取,Agilent公司;标准品磺胺噻唑(ST)、磺胺二甲异恶唑(SIZ)、磺胺多辛(SDM’)、磺胺甲基嘧啶(SM1)、磺胺(SN)、磺胺氯哒嗪(SCP)、磺胺吡啶(SPD)、磺胺甲噻二唑(SMT)、磺胺甲氧哒嗪(SMP)、磺胺嘧啶(SD)、磺胺间甲氧嘧啶(SMM)、磺胺地索辛(SDM)、磺胺甲恶唑(SMZ)、磺胺喹恶啉(SQ)、磺胺二甲嘧啶(SM2)、麻保沙星、洛美沙星、培氟沙星、恩诺沙星、沙拉沙星、氧氟沙星、二氟沙星、诺氟沙星、环丙沙星、达氟沙星、吡哌酸、恶喹酸(奥索利酸)、依诺沙星(氟啶酸)、萘啶酸、氟甲喹、西诺沙星均为 Dr出品;乙腈、甲醇、甲酸(均为HPLC纯试剂),Merck公司;实验用水为Watsons蒸馏水;柠檬酸、磷酸二氢钾、乙二胺四乙酸二钠(EDTA)等其余试剂均为分析纯,广州化学试剂厂;鸡蛋样品,中山市三乡镇白石鸡场。
1.2 仪器与设备
高效液相色谱-三重四级杆串联质谱联用仪G6495A LC-MSD QQQ(配有电喷雾离子源(ESI)及Masshunter B07.01数据处理系统),美国Agilent公司;全自动固相萃取仪,吉尔森公司;冷冻离心机3-30KS,德国Sigma公司;漩涡振荡器,德国IKA公司。
1.3 试验方法
1.3.1 样品提取净化
准确称取5.00 g(精确至0.01 g)鸡蛋样品,加入2%甲酸乙腈8 mL,漩涡震荡1 min,再加入pH 7.0、0.1 mol/L的EDTA缓冲液0.5 mL,漩涡震荡1 min,10000 r/min离心3 min,将上清液转移至QuEChERS EMR-Lipid快速萃取提取管中(此管提前用pH 2.4、0.1 mol/L的乙酸铵溶液5 mL活化),剧烈震荡1 min,10000 r/min离心3 min,将上清液转移至QuEChERS EMR-Lipid反萃取管中,剧烈震荡1 min,10000 r/min离心3 min,吸取1 mL上清液到样品瓶中,50 ℃下氮气吹至近干。
1.3.2 上机测定
浓缩液用0.2%甲酸水:乙腈(4:1)定容至1.0 mL,涡旋混匀,过0.22 µm过滤膜转移至进样瓶中上机检测。
1.3.3 仪器条件
色谱条件:色谱柱:Agilent poroshell EC-C18柱,2.1 mm×100 mm,2.7 µm。
流动相:A相为0.2%甲酸溶液,B相为乙腈溶液;梯度洗脱程序见表1;进样量:5.0 µL。
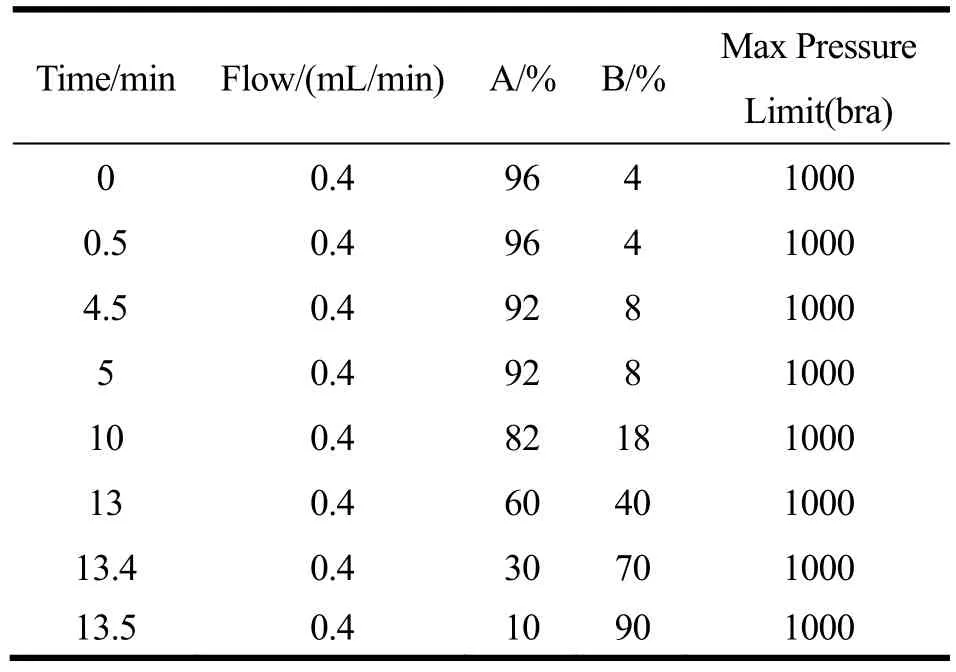
表1 流动相洗脱程序Table 1 Gradient elution program
质谱条件:电喷雾离子源,正离子扫描,动态多反应监测(dynamic MRM),碰撞能量(Fragmentor)为380 V,雾化气压力(ion source gas 1,GS1)为50 psi,毛细管压力(Capillary)为4000 V,干燥气流速(Gas Flow)10 L/min,干燥气温度(Gas Temp)350 ℃,同时优化各物质的质谱检测参数(见表2)。
1.3.4 标准溶液的配制与标准曲线、检测限与定量限测定
根据标准品的实际含量换算标准品的实际称重量,分别准确称取各标准品,置于相应容量瓶中,以10 mL水溶解,并用0.1%甲酸乙腈定容至刻度(其中恶喹酸10 mL水加1.5 mL氨水溶解,再用0.1%甲酸乙腈定容),即成100.00 µg/mL的标准储备液,4 ℃冰箱中避光保存。临用前可将其用流动相稀释至工作浓度标准溶液。
配制一系列浓度的混标溶液,用处理好的空白样品基质,配制0.5、1.0、2.0、5.0、10.0、20.0 ng/mL标准工作溶液,上机检测获得基质标准曲线,数据处理获得线性相关系数。
按3倍信噪比计算方法检测限,10倍信噪比计算方法定量限。
1.3.5 添加回收率、精密度测定
准确称取5.00 g(精确至0.01 g)鸡蛋样品,分别添加标准溶液混合溶液8、16、32 µg/kg浓度,每个添加浓度做6个平行样品,按上述方法进行处理分析,计算回收率和相对标准偏差。
2 结果与分析
2.1 色谱柱选择
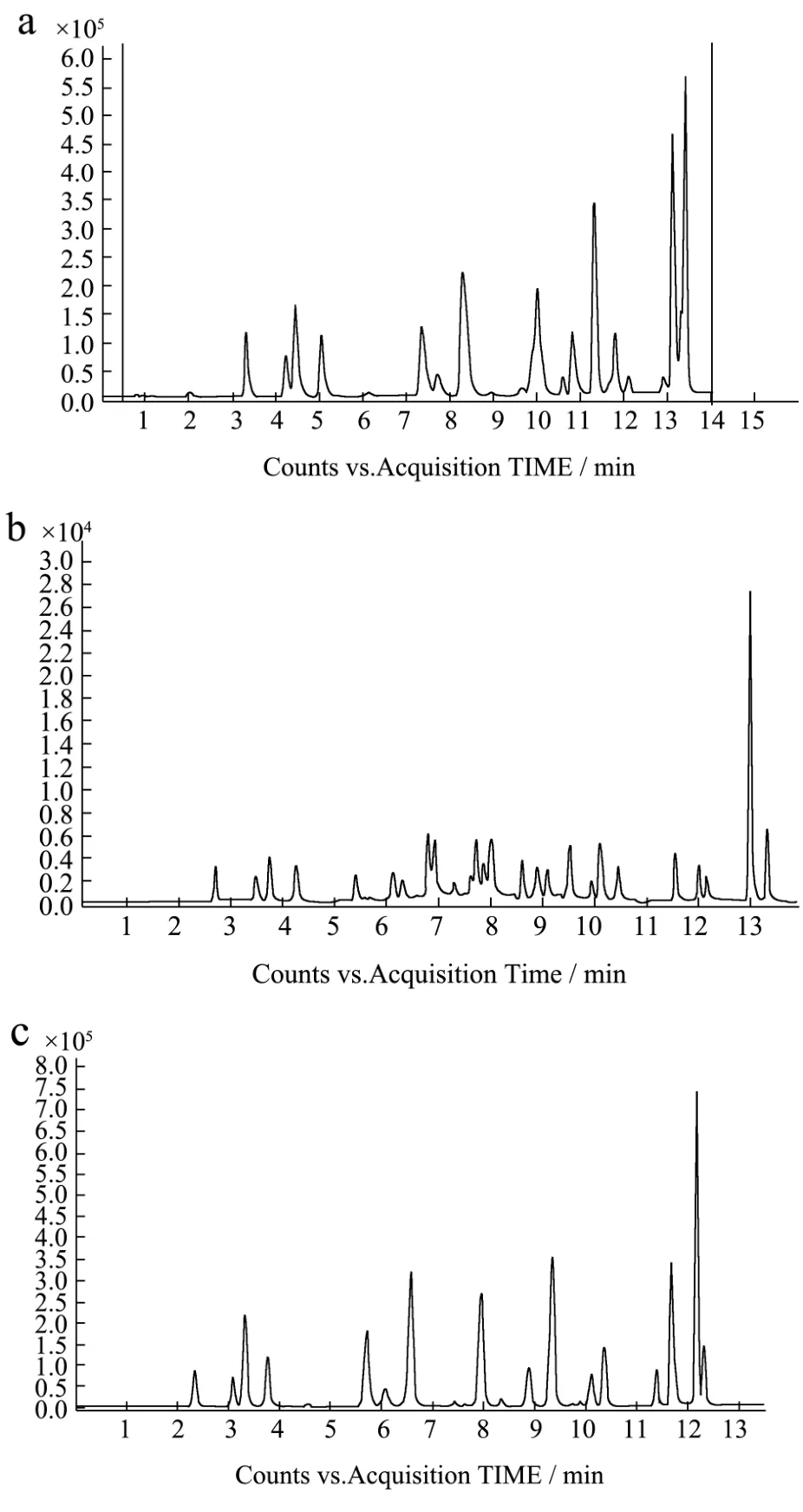
图1 不同色谱柱的选择性离子流图Fig.1 Selective ion flow diagrams of different chromatographic columns
注:X轴为生成时间(m),Y轴为峰高。a.Agilent poroshell SB-C18; b.Agilent poroshell EC-C18; c.Agilent poroshell Phenyl Hexyl
配制 0.1 mg/L的标准溶液混合溶液,分别使用Agilent poroshell EC-C18柱(2.1 mm×100 mm,2.7µm)、Agilent poroshell SB-C18(2.1 mm×100 mm,2.7µm)和 Agilent poroshell Phenyl Hexyl(2.1 mm×100 mm,2.7 µm)柱检测,相同色谱质谱条件下,Agilent poroshell EC-C18柱的响应值和多种药物成分的分离度都比Agilent poroshell SB-C18和Agilent poroshell Phenyl Hexyl的表现好(见图1),所以在以后的试验中,我们选择Agilent poroshell EC -C18柱。
2.2 质谱条件选择

图2 30种抗生素的TIC图Fig.2 Total ion chromatograms of 30 antibiotics
磺胺类药物是人工合成的氨苯磺胺衍生物,基本化学结构为对氨基苯磺酰胺,喹诺酮类药物则都是具有吡啶酮酸共同结构的一组药物,根据两类药物的分子结构性质,我们选择ESI(+)作为电离模式。采用直接进样方式,0.1 mg/L单标溶液进样5.0 µL,正离子扫描模式对各药物进行全扫描,选择响应最佳的准分子离子峰为母离子(parent ion,PI),再对母离子进行二级质谱扫描,得到各个化合物响应值最佳的碎片子离子(daughter ions,DI)。最后再对得到的每种药物的二级质谱的碰撞能量(CE)、电喷雾电压(IS)、雾化气、喷雾压力、毛细管温度和电压等质谱参数进行优化,从而使每种药物的分子离子与特征离子碎片产生的离子强度达到最大时为最佳,使化合物的响应最高(见表2)。在此条件下,各种药物可以得到有效的分离(见图2)。
2.3 色谱条件选择
磺胺类药物和喹诺酮类药物的结构对pH值都有严格的要求。实验比较了 0.2%甲酸-乙腈溶液、0.1%甲酸-乙腈溶液、0.1%甲酸-2 mmoL乙酸铵缓冲液-乙腈溶液、0.1%甲酸-0.05%氨水溶液-乙腈溶液对离子化的影响。结果表明,0.2%甲酸-乙腈溶液体系能使磺胺类药物和喹诺酮类药物在质谱中有更好的响应值,这是由于甲酸可以为阳离子的形成提供必需的质子来源,从而提高其离子化效率。而乙酸铵缓冲液、0.1%甲酸乙腈溶液有明显的拖尾现象,而且响应值低,因此选用 0.2%甲酸-乙腈溶液作为流动相体系。此外,由于柱温太低时流动相粘度大、色谱柱的理论塔板数低、目标物质出峰慢,因此柱温选为40 ℃。
2.4 样品前处理的优化
磺胺类药物和喹诺酮类药物都含有较稳定的结构,微溶于水和甲醇等有机溶液,尤其是在酸性环境下稳定。本实验比较了乙腈、0.1%甲酸乙腈溶液、0.2%甲酸乙腈溶液、0.5%甲酸乙腈溶液、1%甲酸乙腈溶液、2%甲酸乙腈溶液作为提取液提取样品,发现用2%甲酸乙腈溶液提取样品所得的提取液最清,蛋白除去效果最好,磺胺药物的提取效果也较好,所以选择 2%甲酸乙腈溶液作为提取液。喹诺酮类含有吡啶酮酸基本结构,其羧基和酮基极易和金属离子形成络合物,所以单纯 2%甲酸乙腈溶液提取时,喹诺酮类药物的回收率几乎没有,所有我们在样品提取过程中加入乙二胺四乙酸二钠(EDTA)去络合提取管中的金属离子,从而提高喹诺酮类药物的回收率。比较了pH 7.0的0.1 mol/L EDTA、0.2 mol/L EDTA、0.5 mol/L EDTA、1 mol/L EDTA的络合作用,最终发现0.1 mol/L EDTA作为络合剂时喹诺酮类药物的回收率最好。因此在提取过程中,选择2%甲酸乙腈溶液和0.1 mol/L EDTA共同提取样品。
2.5 样品净化
在本实验中,选用PRIME-HLB固相萃取柱、安捷伦QuEChERS快速提取和QuEChERS EMR-Lipid快速提取对样品进行净化。结果表明PRIME-HLB固相萃取柱只对其中的一部分药物有提取,其余药物的提取率低,所以我们选择了安捷伦QuEChERS快速提取进行样品进化。鸡蛋中的蛋白质含量特别高,而脂质干扰是测定食品或生物基质中痕量残留物面临着重要难题,不仅导致分析物灵敏度下降,还会导致 MS维护的需求增加从而缩短仪器和色谱柱寿命,所以去除脂质在痕量检测中尤为重要。我们比较了安捷伦传统 QuEChERS和增强型脂质去除产品(QuEChERS EMR-Lipid)对样品进行净化处理的效果,发现QuEChERS EMR-Lipid对脂质的去除率较好,而且分析物的回收率也较高。所以我们选择 QuEChERS EMR-Lipid进行样品净化过程。
在使用 QuEChERS EMR-Lipid萃取管进行样品净化时,QuEChERS EMR-Lipid快速萃取提取管的活化效果直接影响了检测结果的好坏。本实验比较了纯水、pH 2.4、0.1 mol/L乙酸铵、pH 2.4、0.1 mol/L EDTA、pH 4.0、0.1 mol/L EDTA、pH 7.0、0.1 mol/L EDTA活化提取管,但发现纯水活化时,只有磺胺类药物有回收;pH 4.0、0.1 mol/L EDTA活化时喹诺酮类药物开始有回收,但较低;pH 7.0、0.1 mol/L EDTA活化时喹诺酮类药物回收较好,但磺胺类药物又没有了回收;而pH 2.4、0.1 mol/L EDTA活化时,由于溶液太酸,EDTA有析出现象;而用pH 2.4、0.1 mol/L乙酸铵活化萃取管中,磺胺类药物和喹诺酮类药物均有回收,而且回收率较好,最终选择pH 2.4、0.1 mol/L乙酸铵作为萃取管的活化液进行活化。
2.6 检测方法的线性关系、检测限(LOD)与定量限(LOQ)的确定
配制一系列标准工作溶液,做基质标准曲线进行定量分析,其线性范围(Linear ranges)和相关系数(Correlation coeficiants)见表1。按3倍信噪比计算定为检测限(LOD),10倍信噪比定为定量限(LOQ),结果见表2。
2.7 检测方法回收率和精密度确定
准确称取空白样品,分别添加高、中、低三个浓度标准溶液,每个添加浓度平行测定6次,按1.2所述方法进行处理分析,进行样品分析。药物的回收率较好,回收率范围为50.09%~102.11%之间,精密度也较好,相对标准偏差小于20%,方法满足质量控制要求[30],结果见表3。
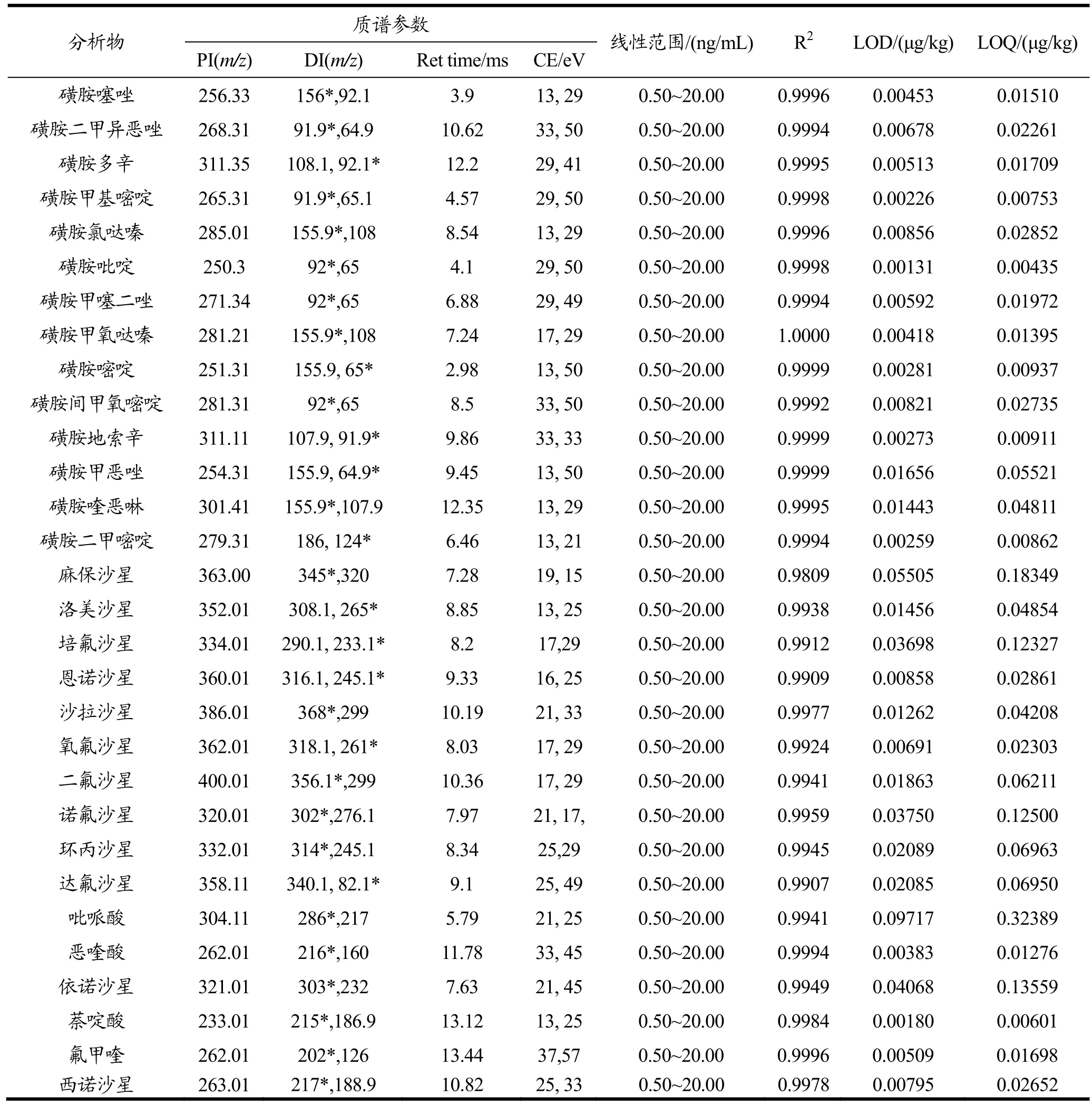
表2 目标分析物的质谱检测参数、工作曲线线性范围、相关系数、检测限和定量限Table 2 Tandem mass parameters, linear ranges, correlation coef fi cients, LOD and LOQ of target analyte drugs
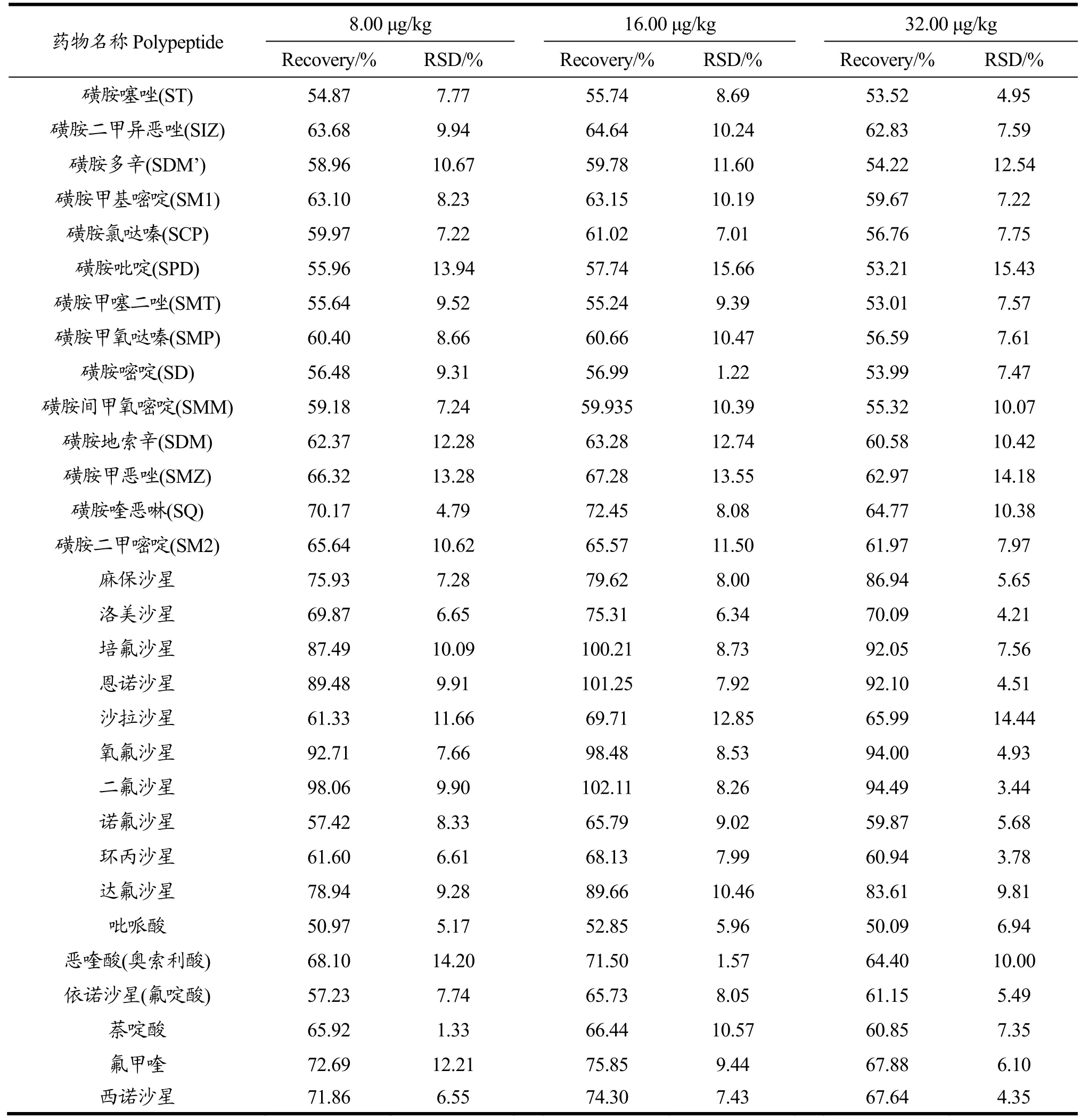
表3 空白样品中30种兽药的加标回收率和相对标准偏差Table 3 Recovery and relative standard deviation for 30 antibiotics in eggs(%, n=6)
3 结论
食品中抗生素残留检测属于较为复杂的痕量组分分析技术,最显著的特点是需要严格的样本前处理步骤,因为前处理的操作直接影响检测结果的准确性和可靠性[31],所以能集采样、萃取、净化、浓缩、预分离、进样于一身的联用和自动化分析等将是未来发展的趋势。而本实验中的检测样品禽蛋中的蛋白质含量较高,在前处理过程中去除脂肪的步骤也非常关键,其脂肪去除的干净程度直接影响了实验的最终结果。但目前报道所用的多数方法通常都会在去除脂质的损失一部分目标物,从而使目标物的回收率受到的影响。所以本实验在样品前处理过程中,采用了创新型安捷伦增强型脂质去除产品(QuEChERS EMR-Lipid)萃取技术净化样品,此技术可针对任意样品前处理产品实现最彻底的脂质去除和分析物回收,不仅提高了方法的灵敏度,还提高了样品的回收率和精密度。而且其操作步骤也比较简单,样品前处理时间也较短,不仅减少了操作过程中的各种误差,而且其还大大节省了样品前处理的时间,适用于大批量产品的快速检测。本试验利用高效液相色谱-三重四级杆串联质谱仪检测禽蛋中磺胺类药物和喹诺酮类药物的多残留含量,不仅具有液相色谱对药物的高效分离特点,也具有质谱对分析物质化学结构的定性分析特性,同时达到了定性和定量的检测目的,特别适用于兽药痕量残留的确证性分析检测,而且在检测结果上大大提高了方法的灵敏度和精确度,所得检测限也比较低,磺胺类药物 检 测 限 为 0.0013~0.0166 μg/kg, 定 量 限0.0044~0.0552 μg/kg;喹诺酮类药物检测限为0.0018~0.0972 μg/kg,定量限 0.0060~0.3239 μg/kg,远远小于欧美国家和我国农业部制定的最高残留限量。与其他液质联用法相比,检测限也有所降低,如王浩[25]等用液相色谱-串联三重四极杆质谱对牛奶中10种磺胺类药物残留,检出限可达到0.5~1.0 μg/L;郭黎明[26]等采用超高效液相色谱-串联质谱在正离子模式下通过多反应监测(MRM)方式同时测定了鸡肝脏组织中3种四环素类药物、10种磺胺类药物以及8种喹诺酮类药物的残留,该方法对测定的21种药物的检出限均为2 μg/kg,定量限均为5 μg/kg。张萍[11]等建立鸡蛋粉中12种磺胺类药物残留量的测定方法,最小检出限在0.002~0.045 μg/g。Schneiderd等使用LC/MC测定了牛和鸡的肝组织中的 sAs,检测限为 0.8~1.7 μg/kg。所以本文建立的QuEChERS EMR-Lipid结合LC-MS/MS检测鸡蛋中多种磺胺类药物和喹诺酮类药物残留含量的检测方法具有高灵敏度、高精密度、前处理操作简便、快速省时等优点,测定的30种兽药残留的含量结果均能满足鸡蛋中多种药物残留检测的限量要求及相应的检测水平,并适用于市场禽蛋中多类兽药的监督抽查工作,在理论和生产实践上都具有重要意义。
猜你喜欢
世界最新医学信息文摘(2022年17期)2022-07-27
核化学与放射化学(2022年2期)2022-04-28
世界最新医学信息文摘(2021年12期)2021-06-09
华东师范大学学报(自然科学版)(2019年2期)2019-06-11
钻井液与完井液(2018年2期)2018-06-13
中国蜂业(2018年4期)2018-05-09
中国卫生标准管理(2015年25期)2016-01-14
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
中国当代医药(2015年36期)2015-03-11
云南畜牧兽医(2015年4期)2015-02-28
