
基于本体参考数据的生物医学本体融合模型研究
2018-10-24 07:59曹春萍
计算机应用与软件 2018年10期
曹春萍 张 政
(上海理工大学光电信息与计算机工程学院 上海 200093)
0 引 言
本体作为一个重要的语义描述系统和存储工具被应用到生物医学领域。目前已开发的生物医学本体是从各知识领域出发,对事物的高度概括和总结,可以提供各领域知识的相对全面、综合性的标准化理解。本体对医学知识概念的高度统一化和标准化,使得相关医学本体经常被应用在疾病研究等方面,提供一致性的知识表示。但是,由于疾病的产生和发展往往具有综合性,涉及到基因、环境、心理、体征表现、疾病相似性关联等多方面的因素,来自单个领域的本体在疾病研究上难以全方面的准确覆盖。为此,将现有本体进行融合就成为当下生物医学中研究的重点。
1 相关工作
目前已有许多针对领域本体融合方法的研究。此类研究主要是从本体自身的语义和结构上的相似性出发,寻找概念间的映射关系,由局部本体向全局本体进行融合的方法。这些融合方法大多通过语义匹配的方式消除本体间的异构。但是,由于不同领域本体的复杂性不同,融合后的本体往往存在局限性大,准确率不高等问题。文献[1]提出一种基于Mediator模式的融合机制,基于本体概念的语义相似度定义了多种本体映射类型,包括直接映射、包含映射和组合映射,并根据映射类型的不同建立了不同融合连接。改进了传统本体概念间进行一对一映射的不足,但缺少了对语义不一致的考虑,并且在映射关联的建立上并没有给出可做倾向性选择的权值参考,不利于融合后本体的应用。文献[2]提出了一种粒化理论的地理本体融合方法。运用了形式概念分析地理本体,再引入粒计算,在不同粒度下通过约简概念格完成本体融合过程。此方法将形式概念分析与粒计算结合用于地理本体融合,打破传统形式背景下的二值局限性,通过粒度划分增加了问题求解的灵活性。文献[3]立足于领域本体的结构特征,分析本体中术语层次结构所包含的语义信息,提出基于属性的计算模型。在本体结构层次中求解概念间的最小不可约集,通过对语义模型引入调节因子,提高了领域概念的融合效率。使用语义度量的方式将概念之间的关系进行加权,同时利用本体层级关系中不同层次概念节点所表达的概念范畴的差别,将概念节点深度对于相似度的影响进行了量化,在一定程度上提高了本体融合的准确性。但在语义距离的计算过程中依然不能避免本体中语义异构带来的影响,并且在多领域本体融合过程中,各本体概念所在层级结构信息对产生跨本体融合连接的作用较小。
还有一些研究借助WordNet等外部词典工具[4-5]或者文档信息进行融合。如基于文献的跨本体融合方法[6],提出了在不同文献中匹配共出现的本体概念对,并从文献的语义描述中挖掘这些术语关联,从而建立本体之间的关联,形成跨领域间本体的融合。由于本体概念存在大量的不同实例,这会导致匹配效果不佳。同时在本体的关联表达上,同样是通过对文档信息的描述分析做语义匹配。由于构成文档的语义描述信息来源复杂,即使进行关键语义抽取后,也可能由于数据来源的专业领域不同而导致得到的关联表达准确度不高。
文献[7]创新性地提出了一种非语义匹配的方式,使用机器学习的方法针对地理本体进行融合。虽然提高了融合效率,但随着融合规模的扩大,概念空间也会异常复杂。本体融合方式主要是建立本体之间的映射关系,通过本体概念、实例及属性之间语义匹配机制和映射方法,实现本体最小元素之间的相似对应关系,从而实现本体的最终融合[8]。因此还有许多研究是通过改进概念间的相似性度量方法来提高本体融合的准确性[9-13]。
生物医学中的本体具有较高的多样性和复杂性,使得通过对生物医学本体的融合以实现知识复用变得困难。对此,不同于上述由局部向全局进行本体融合的一般方式,在生物医学界一般通过对重要本体的融合,为相关问题的解决提供多方位的信息支持。
GO、DO和HPO三个本体在疾病研究上的重要作用是在基因互作、疾病关联、病症表现三个方面提供通用的一致性知识表示,便于不同领域专家对疾病研究达成共同的理解。为此,将其进行融合后得到的标准化信息对病因的多方位研究帮助也会比较大。所以不少学者对其进行了相关研究。文献[6]通过本体概念在文献中的映射关系实现GO与DO的本体融合,但其研究侧重点在于使用丰富的文献信息增加跨本体间的术语关联表达。在文献[16]中,提出了在HPO中基于通路的相似度计算方法,通过与基因网络的结合来进行疾病和致病基因的预测。这类研究中大多是对本体之间的关联进行扩展,并逐步通过相关数据的集成与本体数据进行融合。这种方式没有达到真正意义上的知识融合以形成标准化的共同理解。并且准确率和融合效率一般不高。而目前针对多个核心本体的整体融合研究也相对较少。
本文在GO、DO、HPO三个核心领域本体融合研究中,采用以两两本体融合最终达到整体融合的方式进行。由于融合方法是一致的,所以本文主要以GO与DO本体的融合为例进行论述,同样方法以建立DO与HPO的融合。通过对基因本体GO与疾病本体DO的组织结构和内容描述进行分析,以本体的参考数据来源为切入点,挖掘与当前领域本体关联紧密的相关生物网络数据做非语义匹配。匹配过程中建立了本体注释信息的基因字典树,并改进相关匹配算法,提出了基于本体参考数据的生物医学本体融合模型。
2 基于本体参考数据的本体融合模型设计
2.1 模 型
为了解决由于本体异构导致的“信息孤岛”问题,我们建立跨领域本体之间的融合连接,并在融合过程中尽可能规避繁琐的语义匹配过程,给出了基于本体参考数据的本体融合模型(如图1所示)。在现有本体基础上,引入领域相关性较强的生物网络数据,将传统跨本体间的概念语义匹配问题转化为基因功能相关性表达的问题,简化融合过程,同时提高融合结果的准确性。

图1 模型
基于上述思想,多维度本体融合过程为:
1) 通过本体术语的参考数据源挖掘相关本体注释的生物网络数据。这里使用带有GO与DO本体注释信息的人类基因网络数据(如图2所示)。同理与HPO本体融合过程中使用HPO本体注释的基因功能网络数据。

(a) 基于GO标注的人类基因网络(N1) (b) 人类疾病与其致病基因关联的叙词表(N2)图2 人类基因网络数据
2) 跨本体间术语关联表达使用本体相关生物网络中的基因做非语义匹配建立。这里通过N1与N2中的基因功能相关性表达,确定不同本体术语之间的关联关系,并定性分析和量化这种关联。
2.2 跨本体间术语关联表达
使用不同本体注释的基因功能网络,可以通过基因之间相等或相似的匹配来建立跨本体间的融合连接。并且,基因相关性表达的结果在一定程度上也影响着融合后本体的可检索能力。所以,我们对基因之间的关联从定性和定量两方面进行了分析和研究。定性分析在粗粒度上确定本体之间融合连接的类型,而定量分析则在细粒度上区分关联性的强弱。这样,融合后的本体更具应用性,本体的融合研究才具有意义。
首先,根据基因相关性计算方法不同,我们给出了两种关联表示方法:(1) 显性关联表示;(2) 隐性关联表示。显性关联可以提高不同本体术语关联的准确性和可靠性,隐性关联则允许在一定误差(基因功能网络权重)下可接受的术语关联。这些隐性关联有助于产生新的生物关系猜想。为有价值的生物学发现提供有利基础。
定义1显性关联:找到完全相同的基因使用不同本体术语注释,从而确定不同本体术语间关联。
定义2隐性关联:借助基因功能网络[14],通过基因功能相似性找到不同本体注释术语,从而确定不同本体术语间关联。
其次,借助基因功能网络中的基因功能相似权重系数,分别量化得到的每种关联关系。由于显性关联是通过基因匹配的方式得到,即相同的基因注释不同本体术语,所以通过基因相关性衡量术语相关性的权重系数wx=1。而隐形关联是通过计算不同基因之间的功能相似性来确定术语之间的相似性得来,所以权重系数为计算得到,数值范围在wx∈(0,1)。特别地,当基因不存在基因功能网络NET中时,权重wx=0。gi和gj分别为注释不同本体术语的基因。ti和tj为不同本体中的术语,如下所示:
(1)
2.2.1 显性关联表示:使用本体术语注释的基因集做等价匹配
本体中的每个术语包含一到多个注释基因,大量的基因匹配过程会造成较高的时空开销,所以我们借鉴了AC自动机的思想[15]来降低暴力匹配过程中的时间复杂度。该匹配算法可以对于给定长度为n的文本和模式集合p{p1,p2,…,pm},在O(n)时间复杂度内,找到文本中的所有目标模式,而与模式集合的规模m无关。即我们在建立本体术语间的关联表达时,可以较大程度地忽略掉基因模式的增加给基因匹配效率带来的负担,从而达到我们通过基因的等价匹配来实现跨本体术语关联映射的目的。
具体构造过程可以分为两个阶段:
1) 构造基因字典树。将每种基因逐个字符插入到字典树中,从根节点到叶节点的任意一条路径构成一个完整的基因表示并对应所注释的本体术语表示码。最终构建的字典树如图3所示。
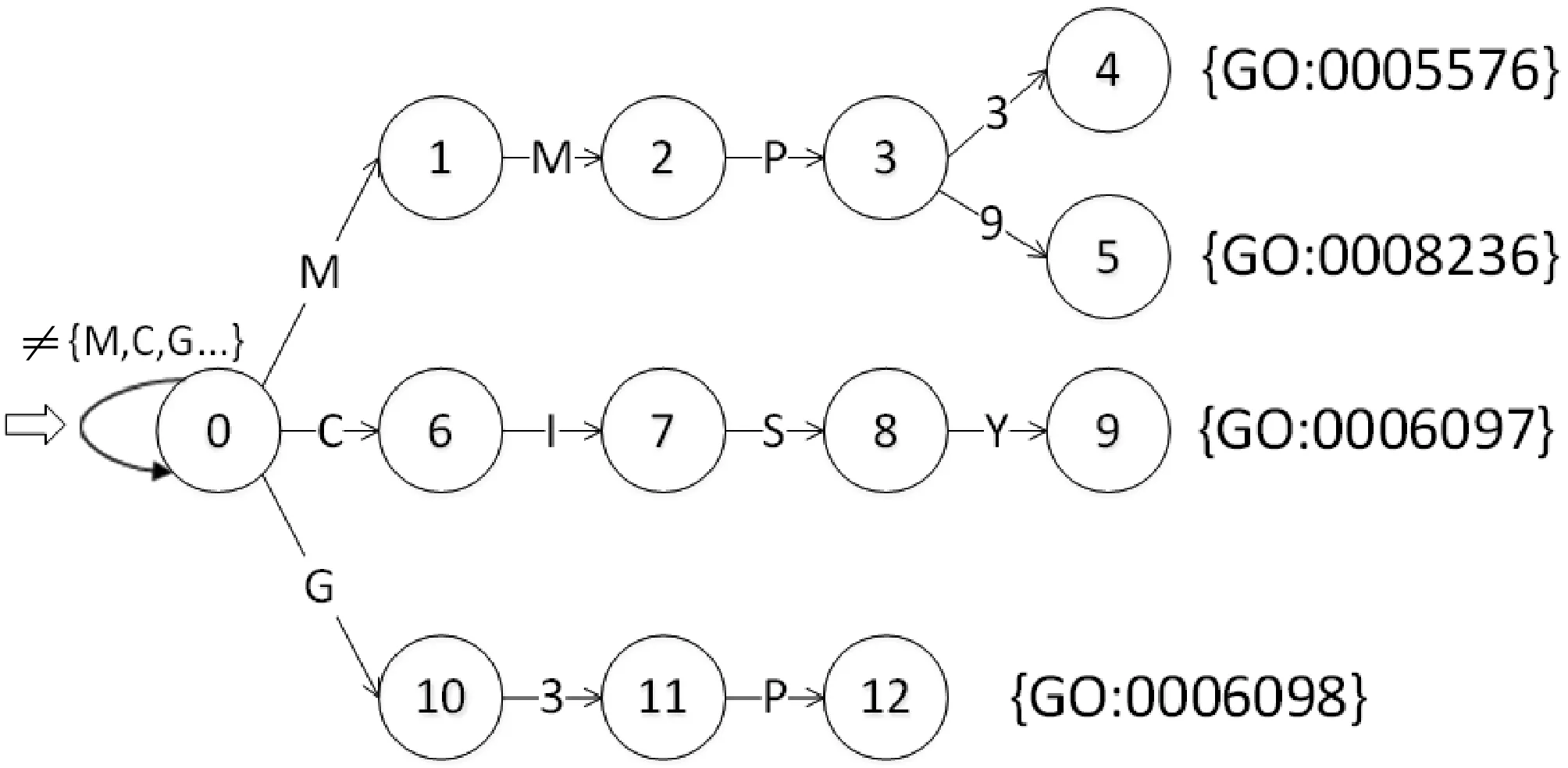
图3 基因字典树(Gene Trie)
字典树构建过程:从唯一的根节点q0开始,从基因集合p={p1,p2,…,pz}中,逐一插入pi(1≤i≤z),并尽可能沿着当前基因pi中字符顺序路径进行,如果pi在状态节点q(q∈Q)中止,在q节点下标记作为pi标识符。如图3所示,我们将用于描述每个基因的GO术语集合(集合中使用GO术语的表示码)作为当前基因模式的标识符;如果在pi中所有字符使用完之前中止,则继续以pi中剩下的字符作为路径进行插入新的状态节点。
完成根节点的转移函数g,如果a∈Σ并且不是根节点q0出来的字符路径上的字符,那么g(0,a)=0。(即q0初始状态)
2) 完成f失配转移函数。这个函数在字典树上以广度优先的方式得到。当计算经过一个字符路径a的状态节点的f函数值时,并假设比当前节点更靠近根节点的f函数值已经计算得到。当发生失配时,回溯到当前状态节点的父亲节点的f函数值所指的状态节点,直到当前状态节点到它的每个直接子节点的字符路径有a的时候,f函数值为此状态节点。如果一直到根节点都没有找到,那f函数值为0。
本文在算法实现中f失配函数的构建中不同于传统AC自动机失配函数在普通连续字符串中的构建方式。因为表示基因的字符是有序一体的,并且基因的字符表示有可能存在包含和被包含的关系,所以在构建失配函数时,不能按最长公共前后缀的方式进行失配转移。如图4所示,当状态7发生失配时,失配指针由父节点指向值为13的状态节点继续匹配可能存在的基因IF。但即使匹配成功,由于从根节点沿字符路径进行匹配的基因CIF只是包含了基因IF,所以不能将IF的GO注释作为CIF的注释。

图4 传统的失配转移
因为是基于基因的等价匹配方式,所以基因字典树通过BFS搜索发生失配时,从基因表示的整体性上考虑,只回溯到当前失配状态节点的父节点(已完成匹配),不再进行当前基因pi的匹配搜索,而是根据pi中下一个字符增加新的状态节点形成新的基因表示以完成第一节阶段基因字典树的动态扩充(如图5所示)。其实质是不同本体参考数据所构成的基因字典树的叠加,最终构成一颗多本体术语注释的基因字典树,增加生物医学领域中其他跨本体术语关联映射的可能。
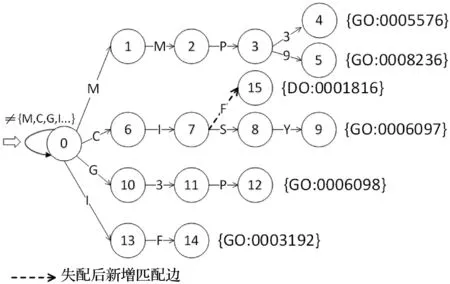
图5 基因字典树动态扩充
实验过程中,改进的匹配算法将GO标注的人类基因网络中的基因在人类疾病和致病基因网络中进行匹配,立起跨本体术语间的显性关联关系。同时,基因字典树的建立也促使了多领域本体进行融合的可能性。
2.2.2 隐性关联表示:使用基于基因功能关联网络的CroGO[14]算法进行匹配关联
定义3基因功能网络(如图6所示):基因功能网络是一个基因功能概率网络。基因功能网络利用了一个改进后的贝叶斯模型,整合了不同类型的生物网络,网络中的节点代表基因,边代表基因之间的功能关联,而边的权重为通过贝叶斯统计模型计算得到的对数似然得分。如果两个基因的对数似然得分为0,表示两基因之间因为功能相关性而匹配的可能性不会比随机匹配的几率高。构建基因功能关联网络的方法,最早由Lee等于2004年提出。
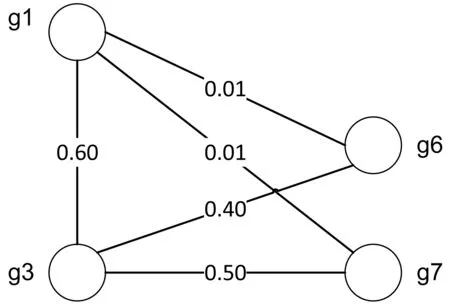
图6 基因功能网络(NET)示例
定义4直接功能距离:在基因网络中任意两个基因节点之间不经过其他任何基因节点而直接相连,则这条边上的权值作为这两个基因节点之间的直接功能距离。
本文使用基因功能网络找到与N2中每种疾病的致病基因关联性强的基因集并借助N1通过CroGO算法计算得到GO与DO术语的隐性关联。使用z-score(标准分数)作为阈值,通过调整合适的阈值大小,确定隐性关联强度。
(2)
式中:x为致病基因到相近基因的功能关联权值,μ表示与致病基因相近基因所有权值的平均值,σ表示所有基因功能关联权值的方差。
不同本体术语隐性关联关系的确定过程同样需要经过两个阶段完成:
1) 使用人类基因功能网络NET,通过基因功能的相似性发现N2中使用DO标注的术语t1对应的致病基因集G1的相近基因集Gsim。
(1) 致病基因gi∈G并且gi存在于基因功能网络NET中,则可以找到与基因gi存在直接功能距离的基因集合Ggi。如图6所示,假如g3为致病基因,则与g3存在直接功能距离的基因集合Gg3={g1,g7,g6}。
(2) 使用标准分筛选基因集和Ggi中功能相关性较强的基因集Gsim。基因功能距离越小则基因功能相关性越低,基因功能距离越大则基因功能相关性越高。使用z-score作为阈值进行筛选,在平均数之上会得到一个正的标准分数,在平均数之下会的到一个负的标准分数。所以,正的标准分数代表基因功能距离大于与当前致病基因gi所有直接功能相关基因功能距离的平均值(即功能相关性越高);负的标准分数代表基因功能距离小于与当前致病基因gi所有直接功能相关基因功能距离的平均值(即功能相关性越低)。
在生物医学上,与致病基因存在直接功能距离,即存在相关性的基因都可能影响当前疾病的产生和发展。即使相似性很低,但存在就有一定的可能性,而这种低的可能性的保留可以针对疾病的深入研究提供更多可考量的方面和探索方向。但本文研究重点在于给出更精准和重要的分子水平描述依据,所以需要通过标准分数筛选出重要的相似基因集。在图6中,通过计算可以得到g3的功能相关性较强的基因集Gsim={g1}。
2) 使用CroGO算法[14]计算本体术语之间的关联权值。
(1) 根据GO本体标注的基因网络找到Gsim基因集的基因子集,记作G2(G2∉∅),每个子集G2唯一对应GO本体中的一条术语t2。
(2) 计算基因G1和G2的关联性,得到t1和t2的术语相似度sim(t1,t2)作为隐性关联强度。基于传统的衡量两个集合关联关系的方法交集比并集。在功能网络NET中,节点表示基因,边表示基因之间的功能相互作用,每条边的权重表示两个基因之间存在的功能相关可能性。两个基因集合G1和G2的功能相关性可以通过公式计算得到。公式如下:
(3)
式中:|X|表示集合X的大小,G1∪G2表示集合G1和G2的并集,f(G1,G2)表示两个集合的差集,由公式计算得到:
(4)
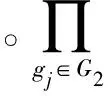
根据基因集合G1和G2计算基因本体术语t1和疾病本体术语t2相似性,计算公式如下所示:
(5)
式中:GSA(G1,G2)由式(3)得到,Gt1和Gt2表示t1和t2所注释的所有基因的组合。
3 实 验
3.1 实验环境与数据集
实验环境配置:算法实现使用Python(v3.6.1)和MATLAB。服务器采用4 GB内存,50 GB硬盘。
为了验证所提方法的表示精度,以及在生物医学上的表现效果。我们分别从KEGG(京都基因与基因组百科全书)、Rectome(人类生物学反应及信号通路数据库)中得到人类疾病与致病基因数据,包含使用GO(基因本体)标注的人类基因9 699条和使用DO(疾病本体)标注的人类疾病1 858种进行实验验证。
本体选择GO基因本体和DO疾病本体作为待融合本体。GO基因本体中选择GO术语总数为42 716条,选择DO本体术语总数为6 878条。由于所选择相关生物网络种类的影响,并不能将本体全部术语信息进行覆盖。这里选择人类相关生物网络做实验验证。
3.2 实验内容及分析
为了验证本文所述的跨本体术语关联算法的计算过程以及本体融合效果,实验过程主要从术语关联精度上进行了验证。并与同样是对GO与DO本体进行融合研究的基于文献的跨本体术语关联算法ARSS[6]进行了比较。
实验一通过本文所述方法,对基因本体术语和疾病本体术语进行关联计算,并得到相应的关联权值对关联术语对进行了定量的分析。通过多样本测试获得的调整的p-value[6]计算所找到的关联,通过比较本文方法与ARSS方法获取的相关术语对是否存在统计上的显著性,来验证术语关联的精度,即验证本体融合方法的精度。
(6)
式中:N是全部的基因数目;M和K分别表示疾病术语和基因本体术语相关的基因数目。X是疾病术语和基因术语共同的基因数目;C(N,K)是从N中选取K的组合。得到的pvalue最终进行了假阳性检验。
如表1所示,分别使用本文方法和ARSS方法对疾病本体和基因本体的术语关联对进行了识别,并从1 000、3 000、5 000(识别的术语对约数)的递增序列来进行具有统计上的显著性的术语对的识别效果的比较。
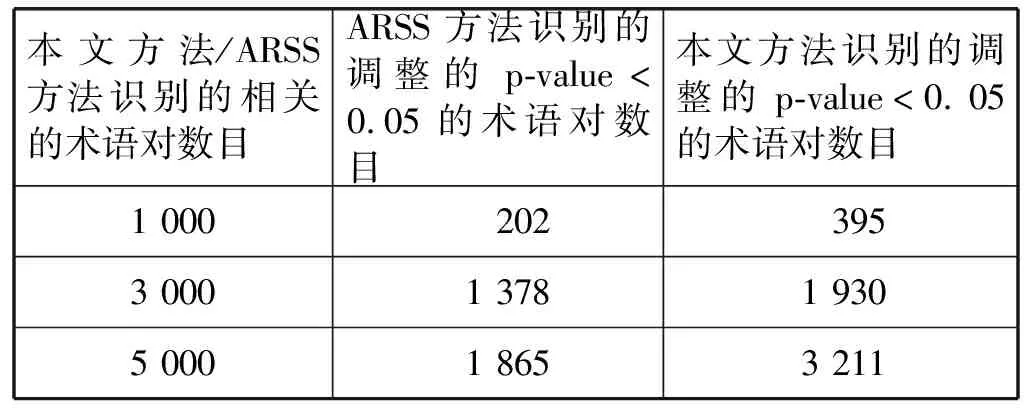
表1 本文方法与ARSS方法识别跨本体术语对数目
通过两种方法在找到的跨本体间的术语关联中,本文方法识别出的具有统计上显著性的术语关联对数目明显高于ARSS识别出的术语对(图7),所以本文方法在识别精度上有一定的提高。并且由于本体参考来源数据对术语对有较高的领域数据贴合性,所以随着术语对的梯度增加,识别出具有统计上的显著性的术语对对数也呈线性增加。

图7 本文方法与ARSS方法对存在统计上的显著性的跨本体术语对的识别验证结果
实验二使用本文所述方法进行跨领域本体融合结果(部分)。
表2和表3中分别给出了不同本体术语间通过显隐性关联表达得到的部分融合连接。以疾病本体中的术语概念“精神分裂症”为例,通过基因功能匹配分别得到与GO和HPO本体中若干术语的关联映射,并给出关联权值W。其中,作用基因是不同本体术语之间映射建立的连接点,权重代表通过基因相关性表达所建立的融合连接强度。相比通过语义匹配建立的关联,通过作用基因的相关性表达得到的关联更具有确定性和倾向性,提高融合后本体的应用能力。连接点表达了融合连接的方式和性质,而连接强度以量化的方式给出了跨本体知识检索中术语关联选择的倾向。

表2 GO、DO本体融合结果(部分)

表3 DO、HPO本体融合结果(部分)
4 结 语
本体为各领域知识提供可共享的理解,在语义网的设计中起到关键性作用,一定程度上决定着语义网中元素具有的语义能力、语义正确性和推理能力,是语义网建设的坚实数据基础。融合后的特定领域本体可以提供多维度的知识理解,形成更广泛的知识表达,是本体在语义网络中发挥重要作用的延伸。本文提出了生物医学领域的本体融合模型,通过挖掘不同本体的来源数据和相关生物网络数据,再通过定性分析,形成领域内多本体之间概念的量化关联,最终达到融合目的。经实验验证,该融合模型具有的一定的准确性和鲁棒性。
猜你喜欢
新世纪智能(数学备考)(2021年9期)2021-11-24
哈哈画报(2021年10期)2021-02-28
当代陕西(2019年15期)2019-09-02
小型微型计算机系统(2019年6期)2019-06-06
学苑创造·A版(2018年11期)2018-02-01
读者(2017年5期)2017-02-15
图书与情报(2013年1期)2013-11-16
卷宗(2013年6期)2013-10-21
中国科技术语(2012年3期)2012-03-20
中国科技术语(2012年3期)2012-03-20
