
乙肝灵胶囊质量标准改进研究
2018-11-09 06:13罗定强袁芬越
中国药业 2018年21期
黄 艳 ,罗定强 ,李 青 ,袁芬越
(1.陕西省食品药品监督检验研究院,陕西 西安 710061; 2.陕西东泰制药有限公司,陕西 咸阳 712000)
乙肝灵胶囊是由大黄、白芍、茵陈、柴胡、贯众、甘草、人参、黄芪8味中药材组方的中药复方制剂,有清热解毒、疏肝健脾的功效,适用于肝气郁滞、湿邪困脾及乙型病毒性肝炎。方中大黄、白芍为君药。大黄苦、寒,泻热通便,利胆逐瘀;白芍苦酸,微寒,养阴和血,柔肝止痛。两者配伍,清利疏养并举。该药品现行质量标准为国家食品药品监督管理局药品标准YBZA17522005-2009Z,其存在部分薄层色谱重现性差、斑点不清晰,且对大黄未进行含量控制的问题。陈光芝等[1]采用高效液相色谱(HPLC)法测定了乙肝灵胶囊中芍药苷含量,而其他药味未见文献报道。为有效控制乙肝灵胶囊的质量,本研究中在对原有鉴别项进行方法学验证的基础上,对大黄、人参的薄层色谱(TLC)法进行了修订,增加甘草的TLC鉴别和大黄中土大黄苷的薄层色谱检查,用HPLC法测定大黄中大黄素和大黄酚的总量,以实现对乙肝灵胶囊质量的有效控制。现报道如下。
1 仪器与试药
1.2 仪器
Waters 2695高效液相色谱仪,2489紫外检测器,Empower工作站(美国 Waters公司);AE204型电子天平(瑞士Mettler Toledo公司)。
1.2 试药
大黄对照药材(批号为 121676-201201),大黄酸对照品(批号为110757-200206),大黄酚对照品(批号为 110796-201520,含量为 99.2%),人参皂苷 Rb1对照品(批号为 110704-201424),人参皂苷 Re对照品(批号为 110754-201324),甘草对照药材(批号为120904-201318),土大黄苷对照品(批号为 110794-201306),大黄素对照品(批号为 110796-200110),均购自中国食品药品检定研究院;乙肝灵胶囊(陕西省东泰制药有限公司,批号分别为 6B01,6B02,6B03);硅胶G薄层板(青岛海洋化工厂分厂);聚酰胺薄膜(浙江省台州市路桥四甲生化塑料厂);甲醇为色谱纯(美国Honeywell公司);水为超纯水;其他试剂均为分析纯,购于国药集团化学试剂有限公司。
2 方法与结果
2.1 TLC鉴别
大黄[2]23,495:取含量测定项下的供试品溶液作为供试品溶液。另取50 mg大黄对照药材,置具塞锥形瓶中,加甲醇10 mL,45 min水浴回流提取,滤过,蒸干滤液,残渣加入8%盐酸溶液10 mL,于沸水浴中加热1 h,冷却至室温,转移溶液置分液漏斗中,用30 mL乙酸乙酯分3次提取,合并乙酸乙酯液,蒸干,残渣用甲醇1 mL溶解,作为对照药材溶液。再取大黄酸对照品、大黄酚对照品,分别加甲醇制成质量浓度为1 g/L的溶液,作为对照品溶液。按处方比例、工艺制备取不含大黄药材的阴性样品0.8 g,按2.3.2项下供试品溶液制备方法制成阴性对照品溶液。照TLC法(2015年版《中国药典(四部》通则0502)试验,吸取供试品溶液和阴性对照品溶液各6 μL,对照药材溶液和对照品溶液各2 μL,分别点于同一硅胶G薄层板上,以石油醚(30~60℃)-甲酸乙酯-甲酸(15∶5∶1)的上层溶液为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视。供试品溶液色谱中,在与大黄对照药材溶液色谱和大黄酸、大黄酚对照品溶液色谱相应位置上显相同颜色的荧光斑点,阴性对照无干扰。详见图1。
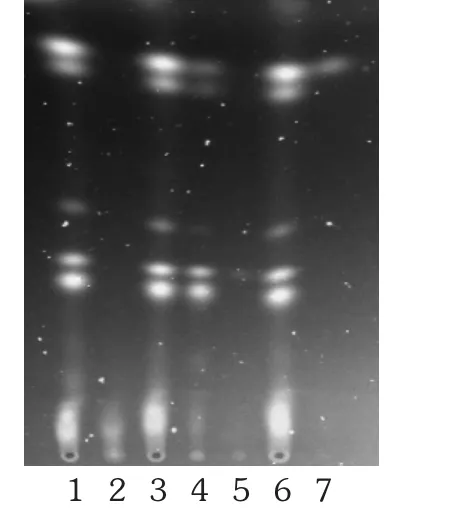
图1 大黄的薄层色谱图
人参[2]468,[3-4]:取本品内容物 6 g,置索氏提取器中,加适量乙醚,加热回流至提取液无色,弃去乙醚液,挥干药渣,再加入100 mL甲醇,加热回流至提取液无色,放冷至室温,提取液蒸干,用30 mL水分次溶解残渣,转移至分液漏斗中,用90 mL水饱和的正丁醇分3次振摇提取,正丁醇提取液合并,用90 mL氨试液分2次洗涤,正丁醇液蒸干,用10 mL水溶解残渣,通过D101型大孔吸附树脂柱(内径为1.5 cm,柱高为12 cm),按50 mL水、40 mL 40%乙醇和60 mL 70%乙醇的顺序洗脱,收集70%乙醇洗脱液,蒸干,用甲醇1 mL溶解残渣,作为供试品溶液。另取人参皂苷Rb1、人参皂苷Re对照品,分别用甲醇制成质量浓度为1 g/L溶液,作为对照品溶液。按处方比例、工艺制备取不含人参药材的阴性样品6 g,同供试品溶液制备方法制成阴性对照品溶液。照TLC法(2015年版《中国药典(四部)》通则0502)试验,上述3种溶液各吸取5 μL,分别点于同一硅胶G薄层板上,以三氯甲烷-甲醇-水(13∶7∶2)10℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,于105℃加热至斑点显色清晰。供试品溶液色谱中,在与人参皂苷Rb1、人参皂苷Re对照品溶液色谱相应位置上显相同颜色的斑点及荧光斑点,阴性对照无干扰。详见图2。
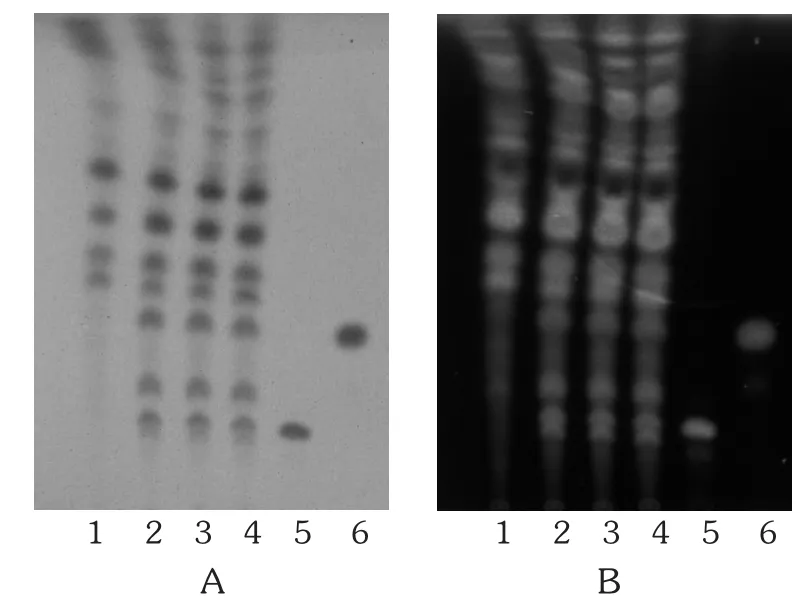
图2 人参的薄层色谱图
甘草[2]86,[5-7]:取本品内容物 1.5 g,置具塞锥形瓶中,量入30 mL甲醇,加热回流1 h,滤过,滤液蒸干,用10 mL水溶解残渣,调节pH至9~10,用60 mL水饱和的正丁醇液分3次提取,弃去正丁醇提取液,水液调节pH至2~3,用60 mL水饱和的正丁醇液分3次提取,提取液合并,用60 mL正丁醇饱和的水分次洗涤2次,蒸干提取液,用甲醇5 mL溶解残渣,作为供试品溶液。另取甘草对照药材0.5 g,同供试品溶液制备方法制成对照药材溶液。再取按处方比例、工艺制备不含甘草药材的阴性样品1.5 g,同法制成阴性对照品溶液。照TLC法(2015年版《中国药典(四部)》通则 0502)试验,取上述3种溶液各2~5 μL,分别点于同一用1%氢氧化钠溶液制备的硅胶G薄层板上,展开剂为乙酸乙酯 -甲酸 -冰醋酸 -水(15∶1∶1∶2),展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,置紫外光灯(365 nm)下检视。供试品溶液色谱中,在与甘草对照药材溶液色谱相应位置上显相同的黄绿色荧光斑点,阴性对照无干扰。详见图3。
2.2 土大黄苷检查[2]23,[8-11]
取本品内容物1 g,置具塞锥形瓶中,加入20 mL甲醇,超声处理 30 min(功率 500 W,频率 40 kHz),滤过,取续滤液作为供试品溶液。另取土大黄苷对照品,加甲醇制成0.1 g/L溶液,作为对照品溶液(临用新制)。按处方比例、工艺制备的不含大黄药材的阴性样品1 g,同供试品溶液制备方法制成阴性对照品溶液。取土大黄替代处方中大黄,然后取按处方比例、工艺所制备的阳性样品1 g,同供试品溶液制备方法制成阳性对照品溶液。照 TLC法(2015年版《中国药典(四部)》通则 0502)试验,上述4种溶液各吸取1 μL,分别点于同一聚酰胺薄层板上,展开剂为三氯甲烷-甲醇-甲酸-水(11∶3∶0.2 ∶0.3),展开,取出,晾干,置紫外光灯(365 nm)下检视。供试品溶液色谱中,在与土大黄苷对照品溶液色谱相应位置上未出现相同颜色的斑点,阴性对照无干扰。阳性对照品溶液色谱中,在与土大黄苷对照品溶液色谱相应位置上出现相同颜色的斑点。详见图4。
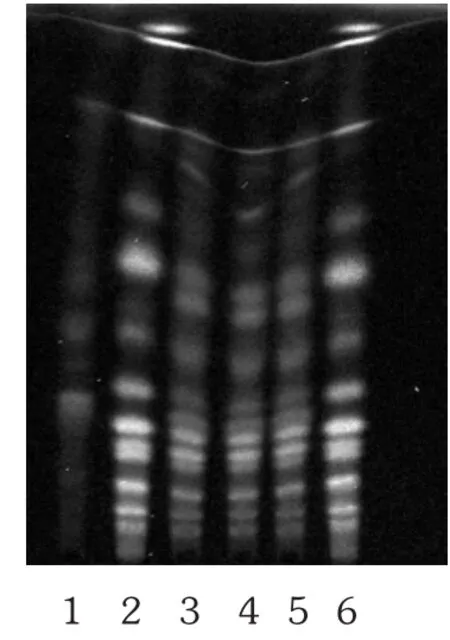
图3 甘草的薄层色谱图
图4 土大黄苷的薄层色谱图
2.3 含量测定[12-15]
2.3.1 色谱条件与系统适用性试验
色谱柱:Agilent 5 TC -C18(2)柱(250 mm ×4.6 mm,5 μm);流动相:甲醇 - 0.1% 磷酸溶液(68 ∶32);流速:1.0 mL /min;检测波长:254 nm;柱温:30 ℃ ;进样量:10 μL。在此条件下,供试品溶液中大黄素色谱峰、大黄酚色谱峰与其他杂质峰基线分离。色谱图见图5。
2.3.2 溶液制备
对照品溶液:取大黄素、大黄酚对照品适量,精密称定,加甲醇制成大黄素质量浓度为10 μg/mL、大黄酚质量浓度为20 μg/mL的混合溶液,即得。
供试品溶液:取样品混匀的内容物约0.8 g,精密称定,置具塞锥形瓶中,精密加入25 mL甲醇,密塞,称定质量,加热回流45 min,冷却至室温,用甲醇补足减失的质量,滤过,精密量取续滤液10 mL,置具塞锥形瓶中,蒸干,冷却,加入8%盐酸溶液10 mL,1 h沸水浴加热,冷却至室温,转移至分液漏斗中,用50 mL乙酸乙酯分次提取5次,合并乙酸乙酯液,蒸干,用甲醇溶解残渣转移至10 mL容量瓶中,定容至刻度,摇匀,滤过,即得。
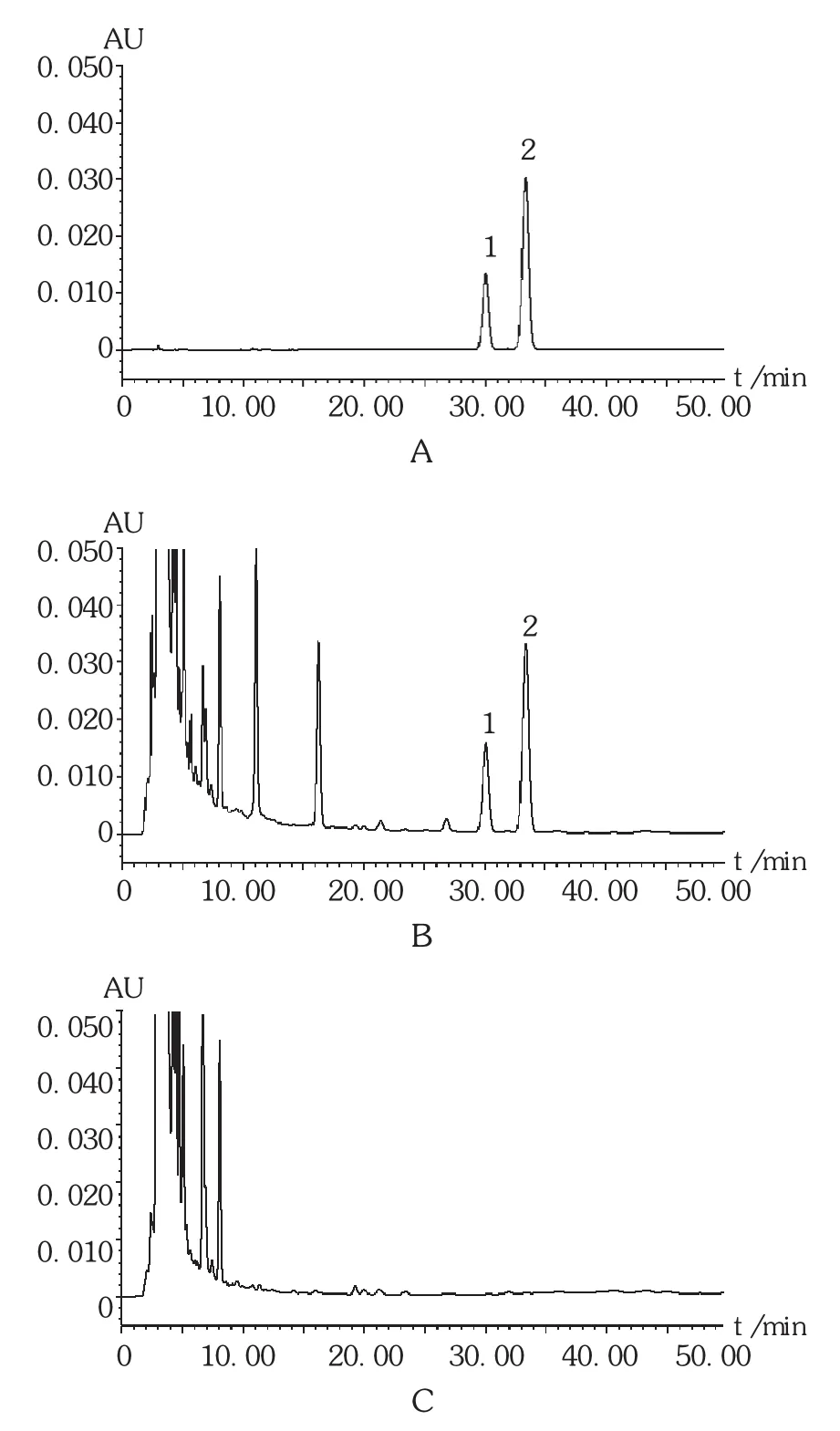
图5 高效液相色谱图
阴性对照品溶液:精密称取按供试品处方比例、工艺制备的不含大黄的阴性样品0.8 g,按供试品溶液制备方法制成,即得。
2.3.3 方法学考察
线性关系考察:精密吸取大黄素(11.70 μg /mL)、大黄酚(19.58 μg /mL)混合对照品溶液 2,5,10,15,20,25 μL,按拟订色谱条件测定,以进样量(X,μg)为横坐标、峰面积积分值(Y)为纵坐标绘制标准曲线,得大黄素回归方程 Y=3 798.28 X+ 271.54,r=1.000 0(n=6);得大黄酚回归方程 Y=5 496.76 X+4 577.04,r=1.000 0(n=6)。结果表明,大黄素和大黄酚进样量分别在 0.023 4~0.292 5 μg 和 0.039 16~0.489 60 μg范围内与峰面积积分值线性关系良好。
精密度试验:精密吸取同一混合对照品溶液10 μL,进样6次。结果大黄素峰面积积分值的 RSD为0.49%(n=6),大黄酚峰面积积分值的 RSD 为 0.85% (n=6),表明仪器精密度良好。
重复性试验:取同一批(批号为6B02)样品,精密称取 6份,每份0.8 g,依法制备供试品溶液,精密吸取10 μL,测定。结果大黄素平均含量为 0.413 2 mg /g,RSD 为 1.57% (n=6),大黄酚平均含量为 0.664 3 mg/g,RSD为1.81%(n=6),表明方法重复性良好。
稳定性试验:取同一供试品溶液,每隔2 h进样1次,记录24 h。结果大黄素峰面积的 RSD为0.34%,大黄酸峰面积的 RSD为0.19%(n=6),表明供试品溶液在24 h内稳定性良好。
加样回收试验:取已知含量的样品(批号为6B02,大黄素含量为 0.413 2 mg /g,大黄酚含量为 0.664 3 mg /g)6份,精密称定,每份0.4 g,分别精密加入25 mL大黄素、大黄酚混合对照品甲醇溶液(大黄素质量浓度为7.656 μg /mL,大黄酚质量浓度为 11.088 μg /mL),依法制备供试品溶液,测定。结果见表1。
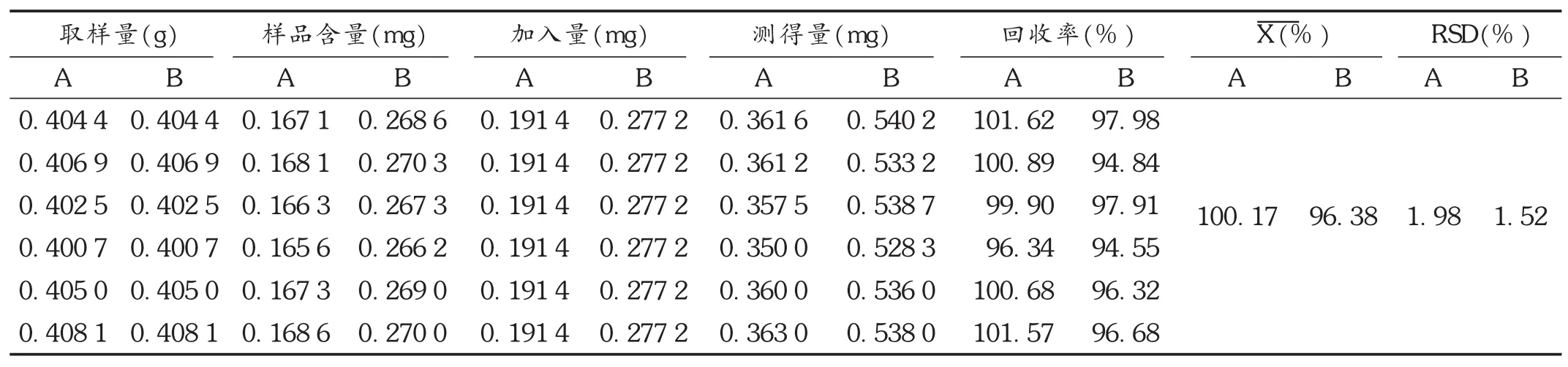
表1 加样回收试验结果(n=6)
2.3.4 样品含量测定
按拟订色谱条件,取3批(批号分别为6B01,6B02,6B03)样品,依法制备供试品溶液,精密吸取 10 μL,注入高效液相色谱仪,测定。结果见表2。
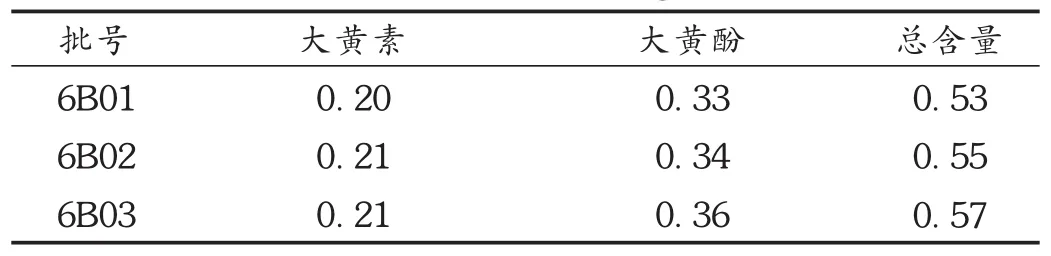
表2 样品含量测定结果(mg/粒,n=2)
3 讨论
现行标准对大黄的薄层鉴别只是以大黄酸、大黄酚对照品为检验指标,且供试品溶液色谱中大黄酸对照品斑点不清晰,对结果的判定有一定影响。本研究中增加了大黄对照药材作为检验指标,以含量测定项下的供试品溶液作为该鉴别项的供试品溶液,优化了薄层鉴别展开系统,考察了不同温度、相对湿度下薄层色谱的展开情况,结果斑点 Rf值适宜,方法耐用性良好。
对大黄素和大黄酚的总量测定,本研究中考察了提取方式、提取时间和水解时间,发现超声处理45 min提取,沸水浴1 h水解,即可提取完全;水解后提取溶剂,比较了三氯甲烷、乙酸乙酯和乙醚3种溶剂,结果发现乙酸乙酯与三氯甲烷提取率相当,其供试品溶液色谱图中预测定峰与其他成分能达到基线分离,因此提取溶剂采用毒性较小的乙酸乙酯。
猜你喜欢
金沙江文艺(2022年1期)2022-02-04
天津大学学报(自然科学与工程技术版)(2021年9期)2021-06-01
健康之家(2021年19期)2021-05-23
中国药学药品知识仓库(2021年18期)2021-02-28
国际放射医学核医学杂志(2021年10期)2021-02-28
化学教与学(2021年12期)2021-02-18
中学生数理化(高中版.高考理化)(2020年1期)2020-11-24
时代邮刊·下半月(2019年8期)2019-09-10
海峡姐妹(2019年8期)2019-09-03
天然产物研究与开发(2019年1期)2019-03-01
