
基于分子标记和高通量测序的基因精细定位
2018-11-20 02:37王茂辉钟春燕罗文龙聂金泉陈志强
广东农业科学 2018年10期
王茂辉 ,钟春燕 ,罗文龙 ,聂金泉 ,郭 涛 ,王 慧 ,陈志强
(1.肇庆市农业科学研究所,广东 肇庆 526000;2.广东省农业科学院蔬菜研究所,广东 广州 510640;3.华南农业大学国家植物航天育种工程技术研究中心,广东 广州 510642)
基因定位主要是基于染色体重组进行连锁分析来定位目标基因,即确定基因在染色体上的位置。DNA分子标记技术的成熟和广泛应用,是近30年来基因定位能够得到快速发展的技术基础,一般以基因组少量分子标记进行的基因定位,需要经过初步定位和精细定位两个阶段。分离群体分组分析法(Bulked Segregation Analysis,BSA)[1]是快速对目标基因进行初步定定位的常用方法,在此基础上利用连锁标记分析隐性单株,就能进一步精细定位目标基因。BSA与多种分子标记可以相结合用于基因定位,常用的分子标记有简单重复序列(Simple Sequence Repeats, SSR)、插入/缺失(Insertion-Deletion, InDel)等[2]。SSR 标记是由2~5个核苷酸为重复单位组成的长达几十个核苷酸的重复序列,由于SSR位点两侧的碱基顺序高度保守,因此可以利用某个两端保守的序列设计一对特异引物,即SSR标记。相对另一个亲本而言,其中一个亲本的基因组中有一定数量的核苷酸插入或缺失,根据这些位点设计的特异性引物就称为InDel标记。
第一代测序技术的标志是1977年Sanger等发明的双脱氧核苷酸末端终止法和Gilbert等发明的化学降解法[3-4],虽然Sanger测序法是直接有效的检测基因突变状态的方法,但也存在一定的不足,如检测基因突变的敏感性较低、实验操作时间长、易产生污染等。第二代测序技术(Next-Generation Sequencing, NGS)相对于以Sanger测序法为代表的第一代测序技术而得名。与Sanger测序相比新一代测序技术共有的突出特征是单次运行产出的序列数据量很大,故而又被通称为高通量测序技术[5],Illumina测序平台是高通量测序技术平台中通量最高。高通量测序技术能快速地一次并行对几十万到几百万条DNA分子进行测序[6]。第三代测序技术是在第二代基础上增加读长、降低成本,并且加快运行速度,其显著特点是单分子测序(Single molecule sequencing,SMRT)[7]。 与Sanger测序法和NGS测序技术相比,SMRT测序具有超长读长、测序周期短、无需模板扩增和直接检测表观修饰位点等特点,为研究人员提供了新选择。高通量测序技术不仅可以进行大规模基因组测序,大大降低了单碱基测序费用,也给基因组学研究带来了更多的新契机。目前高通量测序技术已广泛应用于基因组重测序、小RNAs测序、转录组测序和表观基因组测序等方面[8]。利用极端性状个体混合池进行目标性状基因定位是一种快捷基因定位方法,随着测序技术的革新进步,高通量测序技术越来越多地运用到了基因定位,例如利用BSA与高通量测序结合的BSA-seq(BSA by sequencing)基因定位,并基于BSA分析法开发出了一系列新技术,如MutMap[9]、QTL-seq和MutMap-Gap[10]等。本质上,BSA-seq是利用基因组全局的大量分子标记(几万个以上)进行关联分析,从而快速地进行目标基因定位。
已发现的水稻斑点叶突变体中,大多能提高对稻瘟病及白叶枯病等的抗性并激发病程相关蛋白的表达,发掘新的水稻斑点叶突变体对增强植株抗病性及对抗病反应的认知具有重要的意义。目前报道的水稻斑点叶突变体大多是由于细胞死亡引起的,在第7号染色体上发现的两个斑点叶突变体基因,分别为CRK10[11]和SPL32[12],两个突变体也均出现细胞死亡及早衰现象,而由色素积累导致的斑点叶突变体还少有报道,Spl30突变体出现的红棕色斑点并非是由细胞死亡引起[13]。这一系列关于水稻斑点叶突变体被发掘说明了关于水稻斑点叶突变体研究的重要性,同时相关斑点叶突变体大多涉及细胞死亡以及突变体出现早衰现象,而本研究材料Spl34却未出现细胞死亡和植株早衰。突变体是功能基因组学研究的重要材料,近年来利用水稻突变体进行水稻功能基因组学研究取得重大进展[14-15]。发掘和鉴定各式各样的水稻斑点叶突变体,不仅有助于对水稻品种改良和抗病性研究,同时水稻抗病种质资源的挖掘和研究,对于阐明水稻抗病机制、培育抗病新品种具有重要意义。
1 材料与方法
1.1 试验材料
水稻斑点叶突变体Spl34是从正常绿叶品种粤晶丝苗2号和H4的F2群体中获得的自然突变体,连续多代种植后其突变表型稳定遗传。在前期研究基础上,本研究利用粳稻Francis与Spl34杂交的F2代构建遗传分析和斑点叶基因Spl34(t)精细定位的群体。
1.2 试验方法
1.2.1 遗传分析及群体构建 试验材料种植于华南农业大学校内基地,在2014年晚季以Francis为母本、Spl34为父本,配制杂交组合获得相应F1种子。2015年早季种植F1,2015年晚季种植F2,利用F2分离群体进行斑点叶性状的遗传学分析,以F2(Francis/Spl34)群体作为斑点叶基因精细定位群体。成熟期分别调查父母本、F1和F2群体各个单株的叶上斑点情况,根据斑点有无分为斑点叶和正常绿叶两组,统计两组的植株数,计算分离比例并进行卡平方测验。
1.2.2 基于SSR/InDel标记的基因定位 通过华南农业大学航天育种工程技术中心现有的SSR标记进行亲本多态性分析及群体分析,在初定位区间内通过Gramene数据库对日本晴(粳稻)和9311(籼稻)的基因组序列进行比对,从中筛选InDel位点并设计引物,利用初步定位得到的两个连锁标记InDel-23和InDel-30分析F2(Spl34/Francis)中的无斑点隐性个体,筛选带型为杂合的重组子。利用开发的新标记,对获得的重组子进行筛选,进一步保留其中的杂合个体,通过不断重复新标记筛选交换株,逐步将定位区间缩小。通过水稻基因组在线注释系统RiceGAAS对精细定位区域的基因组序列进行基因注释和功能预测。
1.2.3 基于BSA-seq法的基因定位 混合池测序从F2群体(Francis×Spl34)中,随机选择20个完全没有斑点的个体,取等量叶片混合提取DNA,作为隐性基因极端池;随机选择20个斑点叶性状明显的个体,取等量叶片混合提取DNA,作为显性基因极端池。将极端池的高质量基因组DNA,依次进行DNA片段化、片段纯化、末端修复、3′端加A、连接测序接头、片段大小选择,最终进行PCR扩增形成测序文库(双末端125 bp);建好的文库先进行文库质检,质检合格的文库利用Illumina HiSeq2500进行测序。两个DNA池及两亲本测序产生的原始数据,通过检测确认质量达到要求后比对到日本晴基因组(IRGSP-1.0),进一步检测SNP并计算两个池SNP的等位基因频率(Allele Frequency, AF)[16]。分析测序数据获得候选SNP,通过欧氏距离法(Euclidean distance,ED)关联分析连锁关系,确定候选基因在染色体上的区域。通过水稻基因组在线注释系统RiceGAAS对定位区域的基因组序列进行基因注释和功能预测,对可能的候选基因设计引物进行扩增和测序,比较其在Spl34、分离无斑植株和Francis的序列差异。为了更直观的比较定位区间内的序列差异,将利用IGV(Integrative genomics viewer)对比对的测序数据进行可视化分析,以便于筛选可能的突变基因[17]。
1.2.4 突变体Spl34的单分子测序及数据分析将突变体Spl34提取高质量的基因组DNA,片段化后构建大片段文库(10 kb),利用PacBio RSⅡ进行SMRT测序,原始数据覆盖度15倍以上。对原始数据进行过滤并利用Illumina测序产生的短片段进行碱基校正[18]。校正后的单分子测序数据进行从头组装,生成基因组重叠群(Contigs)。以Spl34基因组一致性序列为参考,将F2混合池的测序数据按相同方法进行比对、SNP检测和关联分析,以确定候选基因在Spl34基因组的定位区域。在此基础上,设计引物扩增,通过Sanger测序比较Spl34和对照的序列差异,并对Spl34和日本晴定位区间序列进行比较分析。
2 结果与分析
2.1 基于SSR/InDel标记的基因定位
2.1.1 遗传分析及定位群体构建 调查Francis/Spl34群体的F1植株表型,发现F1群体植株都表现为斑点叶,说明突变体斑点叶性状受显性基因控制。F2群体植株出现明显的表型分离,分别表现亲本的性状,没有中间类型的植株。随机选取Francis/Spl34的F2群体单株共150株,调查发现其中斑点叶植株为114株,正常绿叶植株为36株,经卡平方测验斑点叶与正常绿叶个体的分离比符合3∶1分离比(χ2=0.436<χ20.05,1=3.840),说明该斑点叶性状受1个显性核基因控制。
2.1.2 基于SSR/InDel的斑点叶基因Spl34(t)精细定位 本研究利用Francis×Spl34杂交的F2群体为定位群体,选取350个F2隐性单株(即正常叶表型)进一步进行精细定位研究。利用现有的第11号染色体上的31对SSR标记对亲本Francis和Spl34进行多态性分析,其中有多态性的SSR标记10对,多态率为32.25%。利用BSA对这10对标记进行连锁分析,发现RM206、RM187和RM254与目标基因存在连锁关系,继续在标记RM206和RM254之间进一步开发InDel分子标记共33个,其中6个有多态性。利用RM206、RM187、RM254以及6个InDel标记对350个正常绿叶的F2单株进行分析,结果标记InDel-19、InDel-23和InDel-30的交换率分别为5.2%、3.3%、10.0%,利用Kosambi作图函数将交换率转换为遗传距离,因此将Spl34(t)基因定位在InDel-23标记附近,遗传距离为3.30 cM(图1A)。InDel-19与InDel-30标记的区间内无法继续开发InDel标记,因此转而开发SSR标记,共找到30个SSR标记,其中6个多态性标记分别为SSR-12、SSR-15、SSR-17、SSR-21、SSR-22和 SSR-30。通过对350个正常绿叶的F2单株进行分析,最终将Spl34(t)基因定位在标记SSR-15与标记SSR-21之间的46.99 kb区间,与SSR-17标记遗传距离为1.71 cM(图1B)。
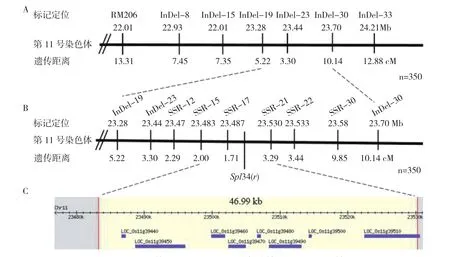
图1 基于SSR/InDel法斑点叶基因Spl34(t)精细定位
2.1.3 定位区间候选基因分析 通过精细定位分析最终将斑点叶基因Spl34(t)定位在SSR-15与SSR-21之间的46.99 kb,通过在线系统RiceGAAS对精细定位区域筛选候选基因,共筛选到8个候选基因(图1C),对8个候选基因进行基因注释及功能预测(表1)。
2.2 基于高通量测序进行BSA-seq的基因定位
2.2.1 测序数据统计和关联分析 对两个具有极端表型差异的亲本构建株系,对子代具有极端表型差异的两个样本池进行等量混合池和全基因组重测序,同时对亲本进行测序,利用BSA法快速有效地寻找与目标性状相关联的基因位点并进行注释。通过BSA-seq对8个水稻建库重测序及2个亲本进行DNA-BSA测序分析,测序数据量结果见表2。

表1 候选基因及其功能注释
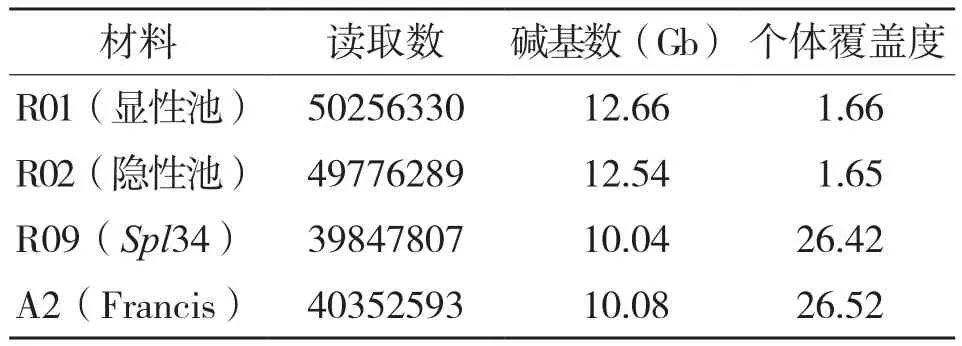
表2 测序数据量汇总
以日本晴为参考序列,将亲本及F2混合池测序数据进行比对和变异检测,并利用ED法分析关联的SNP。在ED法关联分析前,先对SNP进行过滤,首先过滤任一混合池中读取数支持度小于4的位点,得到高质量的可信SNP位点共2 234 370个,并在此基础上识别两混池间差异的位点共1 754 438个。利用ED方法计算关联值,并取原始ED的5次方作为关联值以达到消除背景噪音的功能,然后采用DISTANCE方法对ED值进行拟合,由关联值分布可以看出,第11号染色体上的SNP位点关联效果很好,说明基因定位在第11号染色体(图2,封二)。
根据关联阈值判定,定位区域对应到日本晴基因组坐标为chr11:23 223 211~ 23 791 314,区间大小约568 kb,包含101个转录本(表3)。该定位区间与基于SSR/InDel标记的定位区间部分重叠。但是,由于定位区间内突变体的序列与日本晴基因组差异很大,无法与日本晴进行有效比对,导致混合池SNP等位基因频率与隐性亲本Francis完全一致,因而定位区间无法进一步缩小,由图3(封二)比对到日本晴坐标为 chr:23.43~23.50 Mb。
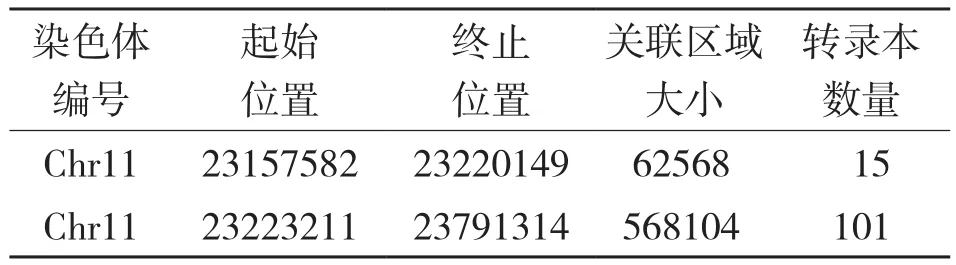
表3 关联区域信息统计
2.2.2 基于单分子测序从头组装序列的关联分析 为了进一步缩小基因Spl34(t)的定位区间,将突变体进行单分子测序和从头组装,并将得到的重叠群为参考进行混合池测序数据的比对、SNP检测和ED关联分析。单分子测序产生数据量为8.54 Gb,进行质量过滤后碱基总量8.29 Gb。对SMRT测序数据进行从头组装,产生4 290个大于10 kb的重叠群,平均长度61.97 kb,累加总长度达到265.86 Mb。将F2混合池的测序数据按相同方法进行比对、SNP检测和关联分析,最终在两个重叠群tig00001409和tig00003011检测到紧密连锁。进一步将两个重叠群的局部序列在日本晴基因组上进行比对,发现两个重叠群分别与日本晴chr11:23.46~23.51 Mb两侧序列高度一致(表4)。两个重叠群长度分别为134.35 kb和92.00 kb,设计引物对Spl34进行扩增后测序,测得两个重叠群之间的间隔(gap)长度7.6 kb。最终,相连的新重叠群序列总长达到233.95 kb,其中Spl34(t)定位区间位于新重叠群区间,大小约为40 kb(图4)。

表4 Spl34重叠群局部序列在日本晴基因组的Blast分析结果
2.2.3 定位区间内序列比较分析 基于在线Multalin(http://multalin.toulouse.inra.fr/multalin/multalin.html)多重序列比对软件,对利用tig00003011末端和tig00001409前端序列为模板扩增的两个重叠群之间7.6 kb区间,比较突变体Spl34和对照在7.6 kb区间序列差异,发现两者没有差异。重叠群tig00003011尾部和重叠群tig00001409前部大小分别为16 989、15 400 bp。根据SMRT测序获得的重叠群序列分别设计11和10对引物后,PCR扩增检测。结果显示,大部分扩增的突变体Spl34与无斑点对照均可扩增出目的片段,而日本晴无法扩增出目的片段,说明日本晴可能存在杂合及非特异扩增现象(图5)。该结果与Spl34重叠群局部序列在日本晴基因组的分析结果一致。
3 结论与讨论
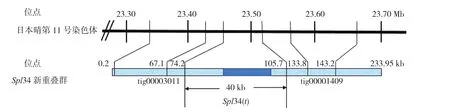
图4 新重叠群在日本晴的基因组坐标
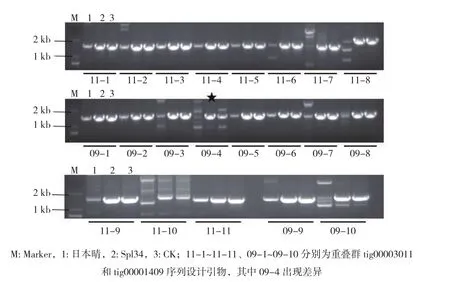
图5 重叠群tig00003011和tig00001409扩增琼脂糖检测结果
本研究遗传分析表明,突变体Spl34的斑点叶表型受单个显性基因Spl34(t)调控,利用Francis×Spl34杂交的F2群体为定位群体,SSR/InDel标记对无斑点叶隐性单株进行连锁分析将斑点叶基因Spl34(t)定位到第11号染色体,定位区间为46.99 kb,并筛选到8个候选基因。在利用SSR/InDel标记对进行精细定位的同时,还通过高通量测序技术进行BSA-seq分析,以确保能够精细定位基因Spl34(t)。高通量测序经过DNA-BSA、SNP过滤、混池间差异识别、ED值关联分析,最后将基因定位在第11号染色体,区间大小约568 kb。为了能够达到精细定位Spl34(t)的目的,继续利用单分子测序技术对Spl34突变体进行测序,对获得的数据辅助校正后从头组装,累加总长度达到265.86 Mb。对F2混合池的测序数据进行BSA关联分析,最终在两个重叠群tig00001409和tig00003011检测到紧密连锁。进一步对Spl34进行扩增后测得两个重叠群之间的间隔长度7.6 kb,最终Spl34(t)定位区间约40 kb。
本研究中试验材料与参考序列差异较大,因此较难开发定位区间里面的高密度标记,很难缩小区间,通过InDel和SSR标记缩小定位区间至46.99 kb,但是突变体Spl34变异区间大小相对于参考序列可能更小。具有类似的研究报道也有很多,如斑点叶突变体hm197[19]的褐色斑点基因定位,突变体Spl21[20]的基因定位等。高通量测序技术的出现则使得区间定位与候选基因识别可直接通过测序完成[21]。然而由于籼稻品种的遗传背景较复杂、差异大等特点,高通量测序与BSA结合的BSA-seq在籼稻中的应用研究还不是很多。本研究通过BSA-seq和单分子测序技术进行基因精细定位,最终定位区间大小约为40 kb。关于此类测序定位的研究还有很多,如在拟南芥的研究中利用少量F2突变体构建混合池测序,并比较SNP比率差从而获得候选区间[22]。
随着高通量测序技术的迅速发展和测序成本的不断降低,越来越多地利用简单快捷的测序手段对基因进行精细定位,例如对突变体基因组直接测序定位、构建混合池测序定位以及遗传分离群体测序构建遗传图谱等,还可以对部分基因组和转录组进行测序定位[23]。本研究基于高通量测序对水稻斑点叶基因Spl34(t)进行精细定位,与分子标记基因定位相比不仅大大降低了时间周期,也提高了基因定位的精确度。传统基因定位分子标记的开发很大程度上依赖于参考序列,本研究中由于突变体Spl34与日本晴序列差异较大导致最后虽然缩短了定位区间。高通量测序大大提高了基因定位效率,随着测序成本的降低、测序技术的发展会有越来越多的基因被发掘及利用。
猜你喜欢
新作文·小学低年级版(2022年3期)2022-08-30
亚热带农业研究(2022年1期)2022-08-08
中国典型病例大全(2022年11期)2022-05-13
智慧少年·故事叮当(2021年5期)2021-08-23
农业科技通讯(2021年1期)2021-03-06
透析与人工器官(2020年1期)2020-11-16
中国农业科技导报(2020年3期)2020-03-15
铁道通信信号(2019年8期)2019-10-10
中国现代中药(2019年5期)2019-07-03
中国食品学报(2019年12期)2019-01-13
