
运动介导AMPK调控线粒体质量控制的机制研究进展
2018-12-13 05:02丁树哲
中国体育科技 2018年6期
张 坦,孙 易,丁树哲
运动介导AMPK调控线粒体质量控制的机制研究进展
张 坦1, 2,孙 易1,2,丁树哲1,2
1. 华东师范大学 青少年健康评价与运动干预教育部重点实验室, 上海 200241; 2. 华东师范大学 体育与健康学院, 上海 200241
作为高度动态化的细胞器,线粒体处于持续动态变化中,其生物发生、融合分裂和自噬之间的平衡对维持线粒体网络稳态至关重要。骨骼肌线粒体的运动性适应包括两个方面,即线粒体数量的增加及线粒体质量(结构与功能)的完善。运动不仅能够促进线粒体生物发生,同时亦可诱导、促进线粒体自噬等逆向适应,其中腺苷酸活化蛋白激酶(Amp activated protein kinase, AMPK)被认为在其中发挥关键作用。本研究综述了运动通过AMPK对线粒体质量控制产生的干预作用及具体分子机制。
运动;线粒体;质量控制;腺苷酸活化蛋白激酶
1 线粒体质量控制概述
线粒体是真核细胞内普遍存在的细胞器,做为细胞生物氧化磷酸化的重要场所,为机体提供大部分能量。然而,线粒体不仅是真核细胞的能量工厂,更是细胞信号转导的调控中心。其中线粒体在氧化磷酸化提供ATP的同时伴随有活性氧(reactive oxygen species, ROS)的产生,过量的ROS则攻击线粒体DNA (mitochondrial DNA, mtDNA)、脂质及蛋白质等生物大分子物质,而且线粒体是ROS首先攻击的靶点。受损线粒体堆积增加进而产生更多的ROS,最终使得氧化还原失衡,形成恶性循环。因此,及时清理受损线粒体并重复利用线粒体内容物,对于维持细胞产生ATP的能力和产生线粒体尤为重要。相应地,机体在长期进化过程中形成了一套完善的机制即线粒体质量控制(mitochondrial quality control, MQC)来确保线粒体数量及质量的相对稳定,其包括线粒体生物发生、动态变化(融合分裂)及自噬三个阶段。首先,两个受损线粒体相遇后经历融合、分裂形成一个功能正常的线粒体和一个线粒体膜电位较低的线粒体,前者进入线粒体循环中重新发挥作用,而后者则不再执行功能,经线粒体自噬途径被选择性清除降解[17](图1)。
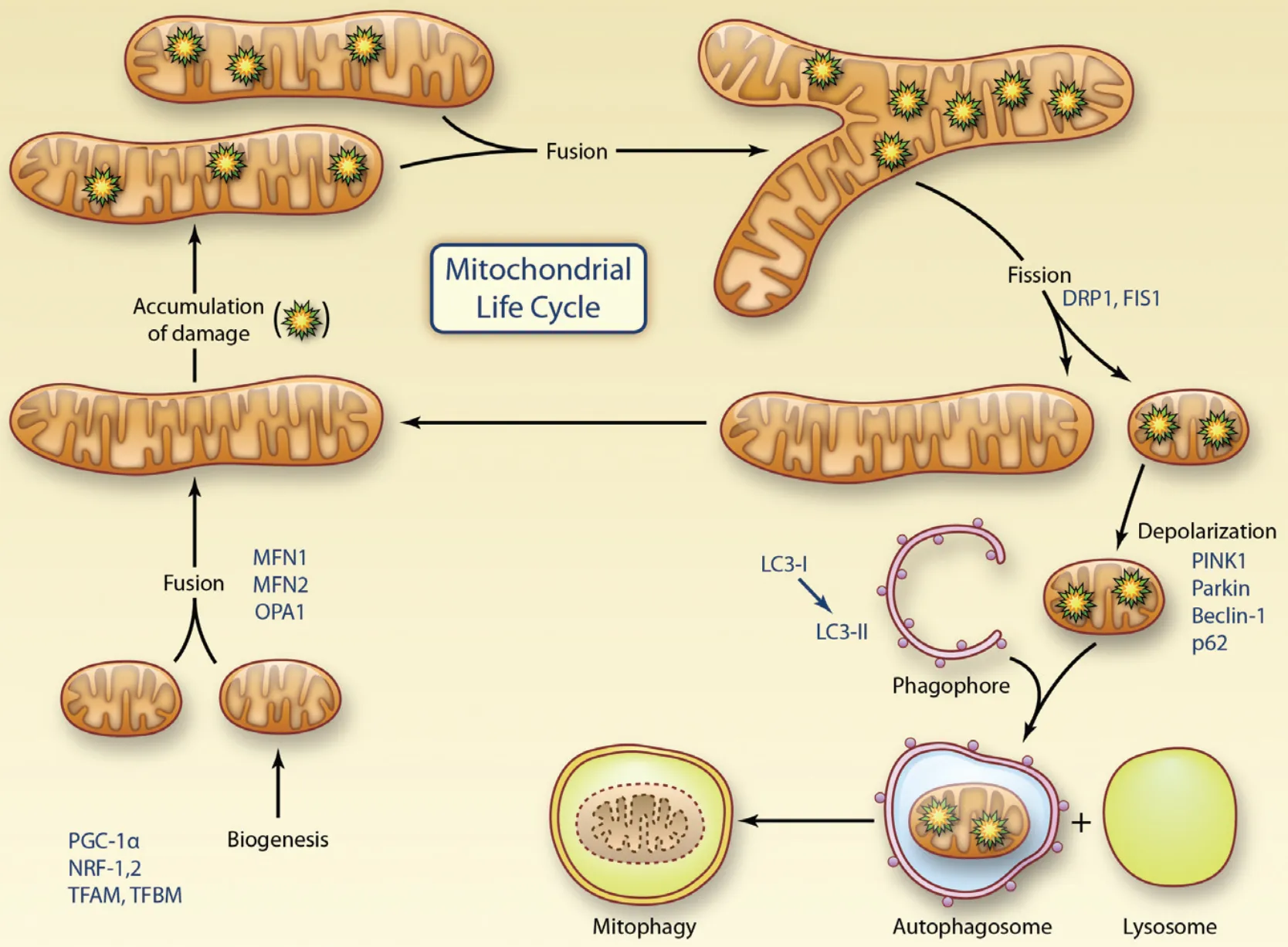
图1 线粒体质量控制示意图[17]
Figure1. Regulation of Mitochondrial Quality Control
注:biogenesis:生物发生;accumulation of damage:损伤累积;fusion:融合;fission:分裂;depoplarization:去极化;phagophore:吞噬泡;lysosome:溶酶体;autophagosome:自噬体;mitophagy:线粒体自噬;mitochondrial life cycle:线粒体生命循环。
2 运动调控线粒体质量控制的分子机制
2.1 运动通过激活AMPK/PGC-1α信号轴促进线粒体生物发生
线粒体生物发生是指细胞中新的线粒体形成的过程。然而需要注意的是,线粒体生物发生过程相对复杂,主要是由于线粒体是半自主细胞器,同时受细胞核基因与线粒体基因的双重调控。已知线粒体中的蛋白质仅有少数是由线粒体基因编码,其余99%的线粒体蛋白质均由细胞核基因编码,所以,线粒体生物发生涉及两个彼此分开的遗传系统。研究表明,线粒体生物发生始于多个细胞核分子事件,其中过氧化物酶体增殖物活化受体γ辅激活蛋白-1α(peroxisome proliferator-activated receptor gamma coactivator-1 alpha, PGC-1α)是目前已知调控线粒体生物发生最为关键的转录因子。在细胞核内,PGC-1α与转录因子相互作用,辅激活核呼吸因子2(nuclear respiratory factor-1/2, NRF-2),同时NRF2又辅激活核呼吸因子1(NRF1),从而上调NRF1和NRF的转录水平。上调的NRFs作为转录因子,进而激活一系列核基因编码的线粒体蛋白基因序列,从而实现对线粒体生物发生过程中所需相关基因的调控[11]。
关于运动与线粒体生物发生的研究始于1967年,Holloszy等人[10]首次报道证实,运动能够促进骨骼肌线粒体增生,即野生型动物肌肉比家养型动物肌肉含有更多的线粒体,同时,长期跑台运动显著增强大鼠骨骼肌的线粒体蛋白表达及酶活性,这一开创性的研究开启了线粒体运动适应的研究之门。随后一系列的研究证实,急性运动与长期运动均可促进骨骼肌中线粒体增生过程中关键蛋白的表达[18,21,34]。而且运动诱导的线粒体生物发生涉及一系列信号通路,其中PGC-1α对于运动诱导的线粒体增生意义重大。一方面,运动应激条件下,细胞质中的PGC-1α移位至细胞核内并通过与NRF1和NRF2共同转录线粒体组成蛋白及线粒体转录因子A(mitochondrial transcription factor A, Tfam),而Tfam是参与线粒体DNA转录、复制以及拟核形成的重要因子。另一方面,运动应激下,PGC-1α移位至线粒体并与Tfam结合形成复合物,从而促进线粒体DNA的转录和复制[27]。由此可知,PGC-1α的移位以及与NRF-1/2、ERR、Tfam的结合是运动调控线粒体增生的关键环节。然而值得注意的是,近期的研究表明,转录因子EB(the transcription factor EB, TFEB)不仅是溶酶体生成的关键调控因子[30],其同样参与调控运动应激条件下线粒体的生物发生,而且不依赖于PGC-1α和PGC-1β[23]。但另有其它研究表明,运动引发的骨骼肌内TFEB表达上调和活性增强依赖于PGC-1α[7],且TFEB可直接与PGC-1α基因的启动子结合进而激活后者的基因表达[29]。这说明目前关于TFEB与PGC-1α、线粒体生成的研究尚未完全明了,仍需进一步的深入探讨。
腺苷酸活化蛋白激酶(AMP activated protein kinase, AMPK)是一种高度保守的丝氨酸/苏氨酸蛋白激酶,是机体重要的能量感受器。诸多研究表明,活化的AMPK可通过上调PGC-1α的活性进而促进线粒体的生物发生[5, 9]。运动、能量限制、肌肉收缩等刺激下,细胞内AMP/ATP比值增加到一定程度可激活AMPK,随后活化的AMPK可直接磷酸化PGC-1α的Thr177和Ser538位点[12],后者进一步移位至细胞核内并与转录因子NRF1和NRF2相互作用,从而激活一系列核基因编码的线粒体蛋白基因序列[11]。其次,PGC-1α亦受翻译后修饰的影响,尤其是乙酰化和磷酸化修饰。如前所述,肌肉收缩引发的能量变化可磷酸化AMPK,后者进而通过高浓度的NAD+激活沉默信息因子2相关酶1(silent mating type information regulation 2 homolog1, Sirt1),随后Sirt1去乙酰化PGC-1α[25]而使其激活,从而触发线粒体生物发生。此外,运动诱导的PGC-1α线粒体移位同样依赖于AMPK[32]。值得一提的是,Rabinovitch等人[26]最新的研究结果表明,线粒体ROS(mROS)是一种生理性的AMPK激活剂,且mROS对于激活AMPK及其下游一系列级联反应,如PGC-1α的活化和细胞自噬关键因子ULK1(uncoordinated -51-like-kinase 1)的磷酸化均是必需的。由上可知,AMPK作为关键信号分子,在运动调控的线粒体生物发生进程中具有不可替代的中心调控作用,且运动应激条件下引发的机体能量代谢的变化及mROS的生成均可能激活AMPK,进而促进线粒体生物发生。
2.2 运动可能通过激活AMPK/MFF信号通路促进线粒体分裂
作为细胞内重要的供能细胞器,线粒体功能是机体生理病理变化中的关键指证。在正常生理状态下,细胞内线粒体处于动态变化中,包括线粒体形态和结构的变化、线粒体在细胞内分布的变化、单个线粒体的分裂再生与线粒体之间的相互融合。线粒体的动态变化包括线粒体的融合与分裂,且融合分裂事件主要由多种蛋白质精确调控完成。参与线粒体融合的关键分子有线粒体融合蛋白1/2(mitofusin1, Mfn1; mitofusin2, Mfn2)、视神经萎缩相关蛋白1(optic atrophy, OPA1)等,其中线粒体外膜(outer mitochondrial membrane, OMM)的融合由Mfn1/2介导,线粒体内膜(inner mitochondrial membrane, IMM)的融合则由OPA1介导;参与线粒体分裂的关键分子有动态相关蛋白1(dynamin-related protein 1, DRP1)和线粒体分裂蛋白1(mitochondrial fission 1, Fis1),其中OMM的分裂由DRP1介导调控,Fis1或线粒体分裂因子-1(mitochondrial fission factor, MFF)可募集DRP1至受损线粒体。进一步研究显示,DRP1定位至OMM依赖于Fis1,但也有研究发现,哺乳动物敲除Fis1并未影响DRP1的定位[2],这说明Fis1对于线粒体分裂可能是非必需的,而其它关键性分子,如MFF对于DRP1的定位及线粒体分裂则起关键作用[39]。
然而,目前关于运动对线粒体动态变化影响的研究并不多,且多聚焦于不同方式运动对线粒体融合分裂相关基因表达的影响层面。早期研究表明,一次性跑台运动可显著降低SD大鼠及人体骨骼肌中线粒体融合相关基因Mfn1、Mfn2的表达[1,4],但近期有研究显示,一次性抗阻运动对线粒体融合分裂基因的表达均无影响,同时,长期抗阻运动或抗阻运动结合热量限制则均能显著增加融合基因Mfn1、Mfn2、OPA1的表达[15,16],这说明不同方式、不同强度运动介导的线粒体融合的分子机制可能不同。此外,与线粒体融合对运动的应激反应不同,一次性跑台运动及长期跑台运动均显著上调线粒体分裂相关基因Fis1的表达[1,4]。
以上研究结果说明,目前关于运动与线粒体融合分裂的研究有限,尤其是关于运动调控线粒体融合分裂的分子机制的研究较少。值得注意的是,Toyama等人[33, 38]最近研究发现,活化状态的AMPK能够磷酸化MFF Ser155/Ser172位点,进而募集DRP1定位至OMM,随后开启线粒体分裂,而且MFF的磷酸化缺失可抑制线粒体分裂,阻碍线粒体自噬,这说明AMPK诱导的MFF磷酸化对于线粒体分裂及线粒体自噬是必需的。因此推测,运动可能通过激活AMPK,磷酸化MFF,进而促进线粒体分裂及随后的线粒体自噬。
2.3 运动通过激活AMPK/mTOR/ULK1信号途径促进线粒体自噬
线粒体自噬是指在氧化损伤、营养缺乏、细胞衰老等应激条件下,细胞内的线粒体发生去极化损伤,其中两个受损线粒体经融合分裂过程形成一个功能正常的“健康线粒体”和一个线粒体膜电位(mitochondrial membrane potential, MMP)较低的线粒体,后者被特异性包裹进入自噬体中并与溶酶体结合,从而完成损伤线粒体的降解清除,维持线粒体稳态。关于损伤线粒体如何被自噬体识别,目前研究普遍认为PINK1/Parkin(PTEN-induced putative kinase protein1/Parkin)、Bnip3(BCL2 and adenovirus E1B 19kDa protein-interacting protein 3)/ Nix(NIP3-like protein X, NIX, 又称BNIP3L)以及FUNDC1是介导线粒体自噬的关键通路,其中PINK1/Parkin是最为经典的线粒体自噬途径。PINK1是一种丝氨酸/苏氨酸蛋白激酶,正常生理条件下,PINK1合成后不断转移至线粒体并与线粒体外膜转运酶(translocase of the outer membrane, TOM)结合形成一个700 kDa的复合物进而进入线粒体膜间隙[19],随后又与线粒体内膜转运酶(translocase of the inner membrane, TIM)作用,并被内膜菱状蛋白(presenilin-associated Rhomboid-like protein, PARL)迅速降解[13]。因此健康线粒体中PINK1的含量较低,其自噬活性处于基础水平。在损伤、衰老等应激状态下,线粒体膜电位下降,PINK1不再向内膜转运及与TOM结合,而是定位于线粒体外膜并从细胞质中募集Parkin至损伤线粒体。Parkin是一个E3泛素连接酶,可级联泛素化下游的一系列线粒体蛋白,如线粒体装配调节因子(mitochondrial assembly regulatory factor, MARF)、Mfn1/Mfn2和电压依赖性阳离子通道蛋白1(voltage-dependent anion selective channel protein , VDAC)等。最终抑制受损线粒体的流动性及其与正常线粒体的融合,促进自噬体对受损线粒体的特异性识别并包裹,使得受损线粒体得以清除。与PINK1/Parkin不同,Bnip3/Nix途径介导调控哺乳动物红细胞中线粒体的降解,一方面,Nix可与Bcl-2竞争性结合来解离Bcl-2/Beclin1复合物,诱发自噬。另一方面,Nix不仅参与募集Parkin至受损线粒体,还可与LC3蛋白直接结合并招募LC3至损伤线粒体,进而激活线粒体自噬[24]。此外,2012年,Liu等人[22]首次鉴定出了一种在缺氧条件下参与调控哺乳动物线粒体自噬的线粒体膜蛋白FUNDC1。在低氧条件下,蛋白激酶Src磷酸化FUNDC1 的LIR Thy18位点,抑制FUNDC1与LC3的相互作用,进而下调线粒体自噬活性。但同时低氧刺激亦能募集ULK1至受损线粒体,ULK1进而磷酸化FUNDC1的Ser17位点,诱导线粒体自噬[35],这说明低氧诱导的线粒体自噬途径并非单一,而可能是多条通路共同作用的结果。此外,Chen等人[3]近期的研究结果表明,FUNDC1不仅通过与LC3b结合进而激活线粒体自噬,它还可与DRP1和OPA1相互作用进而调控线粒体融合或分裂。
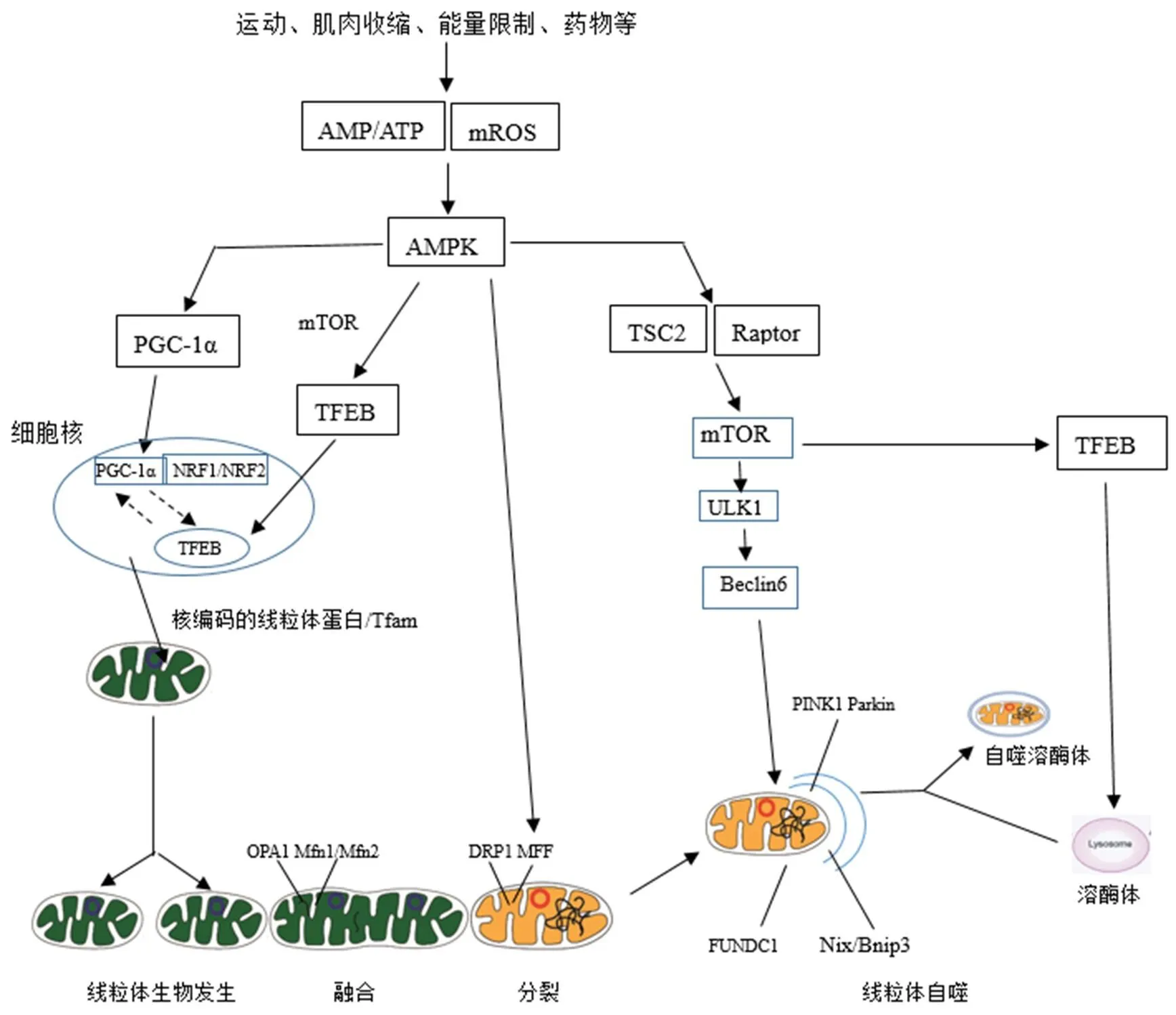
图2 AMPK介导运动调控线粒体质量控制的分子机制示意图[5]
Figure2. The Molecular Mechanisms of Exercise-induced AMPKRegulating Mitochondrial Quality Control
注:实线箭头代表已经被证实的通路,虚线箭头代表运动介导的可能通路。
以往关于线粒体的研究主要集中于线粒体生成等正向适应领域,对于线粒体自噬这一“逆向适应”领域的报道较少。随着研究的深入,发现运动不仅促进线粒体生成(正向适应),也能激活线粒体自噬。诸多研究显示,在线粒体自噬的进程中,一系列级联放大信号事件参与其中。迄今为止,研究较为深入的是AMPK/哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin, mTOR)/ ULK1途径。在正常生理状态下,mTOR磷酸化ULK1 Ser757位点,阻碍AMPK与ULK1的相互作用,进而降低ULK1的活性并抑制其与Atg13、FIP200结合形成ULK1-Atg13-FIP200复合物,从而确保细胞自噬处于基础水平[14]。而运动、能量限制、白藜芦醇、AICAR等应激因素可激活AMPK进而促进线粒体自噬。活化的AMPK可磷酸化TSC-2(tuberous sclerosis complex 2),下调Rheb-GTP,从而抑制mTORC1的活性[31];也可磷酸化接头蛋白Raptor,增加Raptor与14-3-3蛋白的结合,从而阻碍Raptor与mTOR或mTOR底物的结合[8],解除mTOR对ULK1 Ser757位点的磷酸化抑制作用,诱导ULK1与AMPK结合,随后AMPK磷酸化ULK1 Ser317/Ser467/Ser555/ Ser637/Ser777/Thr574位点,增强ULK1活性并促进ULK1-Atg13-FIP200复合体的形成[14,20]。在ULK1的诸多磷酸化位点中,Ser555位点被认为是AMPK的主要磷酸化靶点[6]。进一步的研究结果也证实,AMPK激活剂AICAR能够上调ULK1 Ser555的磷酸化水平,解除mTOR与ULK1-Atg13-FIP200复合体之间的作用,促进线粒体自噬。此外,有研究显示,AMPK不仅能够通过磷酸化TSC2和Raptor途径抑制mTOR活性,其还可通过抑制mTOR向溶酶体的定位来激活线粒体自噬[28]。值得注意的是,近期有研究证实,AMPK被激活后通过抑制mTOR可引发TFEB的去磷酸化,随后TFEB移位至细胞核内并参与调控一系列与溶酶体活性和自噬密切相关的基因表达[9, 37]。
以上研究结果说明,运动、能量限制、白藜芦醇、AICAR等应激因素可激活AMPK,活化状态的AMPK不仅通过磷酸化TSC2和Raptor,抑制mTOR的活性,增加AMPK与ULK1的相互作用,上调ULK1的活性,激活线粒体自噬;同时,AMPK还可抑制mTOR向溶酶体的定位进而上调线粒体自噬。
3 研究小结
运动对线粒体质量控制具有关键调控作用[5,36],其中AMPK是运动调控线粒体质量控制的汇聚点。首先,运动通过激活AMPK/PGC-1α途径增加线粒体生物发生;其次,运动可通过上调AMPK/MFF信号轴促进线粒体分裂;此外,运动亦可通过诱导AMPK/mTOR/ULK1通路激活线粒体自噬途径。
然而,关于运动与线粒体质量调控的研究仍存在一些亟待解决的关键问题:1)AMPK是调控线粒体质量控制机制的枢纽,但AMPK是否存在于线粒体中目前仍旧未知。鉴于线粒体是机体能量产生的重要场所,而AMPK又是对能量变化极其敏感的感受器,因此推测AMPK可能定位于线粒体中,但该假设亟需进一步的研究证实[9]。2)PINK1/Parkin是目前已知调控线粒体自噬最为经典的途径,但AMPK及其下游靶基因是否参与调控PINK1/Parkin途径尚未明确。基于羰基氰化物间氯苯腙(carbonyl cyanide 3-chlorophenylhydrazone, CCCP)不仅是AMPK的激活剂,同时还可诱导PINK1/Parkin调控的线粒体途径,因此推测AMPK及其下游靶基因可能参与调控PINK1/Parkin途径。3)已有研究表明mROS是AMPK的激活剂,对于激活AMPK及其下游一系列级联反应不可或缺。但运动应激引发的mROS生成增多能否通过激活AMPK进而调控其下游的一系列通路仍缺乏直接证据。4)已知活化的AMPK可引发TFEB移位至细胞核内并参与调控自噬相关基因的表达,且TFEB亦被证实参与调控运动应激下线粒体的生成。但目前鲜见研究报道AMPK能否通过促进TFEB的核转位,进而同时调控线粒体生物发生和线粒体自噬。除此之外,TFEB调控的线粒体生成途径是否依赖于PGC-1α也尚未有定论。虽然目前关于运动与线粒体质量控制的研究仍存在较多空白,但不可否认的是,以上问题的解决将有助于人类更加充分地认识运动调控线粒体质量控制的分子机制,并为今后线粒体相关疾病的治疗提供理论依据。
[1] BORI Z, ZHAO Z, KOLTAI E,. The effects of aging, physical training, and a single bout of exercise on mitochondrial protein expression in human skeletal muscle[J]. Exp Gerontol, 2012, 47(6): 417-424.
[2] CHAN D C. Mitochondria: dynamic organelles in disease, aging, and development[J]. Cell, 2006, 125(7): 1241-1252.
[3] CHEN M, CHEN Z, WANG Y,. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy[J]. Autophagy, 2016, 12(4): 689-702.
[4] DING H, JIANG N, LIU H,. Response of mitochondrial fusion and fission protein gene expression to exercise in rat skeletal muscle[J]. Biochim Biophys Acta, 2010,1800(3):250-256.
[5] DRAKE J C, WILSON R J, YAN Z. Molecular mechanisms for mitochondrial adaptation to exercise training in skeletal muscle[J]. FASEB J, 2016, 30(1): 13-22.
[6] EGAN D F, SHACKELFORD D B, MIHAYLOVA M M,. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy[J]. Sci, 2011, 331(6016): 456-461.
[7] ERLICH A T, BROWNLEE D M, BEYFUSS K,. Exercise induces TFEB expression and activity in skeletal muscle in a PGC-1alpha-dependent manner[J]. Am J Physiol Cell Physiol, 2017: 162.
[8] GWINN D M, SHACKELFORD D B, EGAN D F,. AMPK phosphorylation of raptor mediates a metabolic checkpoint[J]. Mol Cell, 2008, 30(2): 214-226.
[9] HERZIG S, SHAW R J. AMPK: guardian of metabolism and mitochondrial homeostasis[J]. Nat Rev Mol Cell Biol, 2018, 19(2): 121-135.
[10] HOLLOSZY J O. Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle[J]. J Biol Chem, 1967, 242(9): 2278-2282.
[11] HOOD D A, TRYON L D, CARTER H N,. Unravelling the mechanisms regulating muscle mitochondrial biogenesis[J]. Biochem J, 2016, 473(15): 2295-2314.
[12] JAGER S, HANDSCHIN C, ST-PIERRE J,. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha[J]. Proc Natl Acad Sci USA, 2007, 104(29): 12017-12022.
[13] JIN S M, LAZAROU M, WANG C,. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL[J]. J Cell Biol, 2010, 191(5): 933-942.
[14] KIM J, KUNDU M, VIOLLET B,. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1[J]. Nat Cell Biol, 2011, 13(2): 132-141.
[15] KITAOKA Y, NAKAZATO K, OGASAWARA R. Combined effects of resistance training and calorie restriction on mitochondrial fusion and fission proteins in rat skeletal muscle[J]. J Appl Physiol (1985), 2016, 121(3): 806-810.
[16] KITAOKA Y, OGASAWARA R, TAMURA Y,. Effect of electrical stimulation-induced resistance exercise on mitochondrial fission and fusion proteins in rat skeletal muscle[J]. Appl Physiol Nutr Metab, 2015, 40(11): 1137-1142.
[17] KLUGE M A, FETTERMAN J L, VITA J A. Mitochondria and endothelial function[J]. Circ Res, 2013, 112(8): 1171-1188.
[18] KONOPKA A R, SUER M K, WOLFF C A,. Markers of human skeletal muscle mitochondrial biogenesis and quality control: effects of age and aerobic exercise training[J]. J Gerontol A Biol Sci Med Sci, 2014, 69(4): 371-378.
[19] LAZAROU M, JIN S M, KANE L A,. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin[J]. Dev Cell, 2012, 22(2): 320-333.
[20] LIN M G, HURLEY J H. Structure and function of the ULK1 complex in autophagy[J]. Curr Opin Cell Biol, 2016, 39: 61-68.
[21] LITTLE J P, SAFDAR A, BISHOP D,. An acute bout of high-intensity interval training increases the nuclear abundance of PGC-1alpha and activates mitochondrial biogenesis in human skeletal muscle[J]. Am J Physiol Regul Integr Comp Physiol, 2011, 300(6): R1303-R1310.
[22] LIU L, FENG D, CHEN G,. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells[J]. Nat Cell Biol, 2012, 14(2): 177-185.
[23] MANSUETO G, ARMANI A, VISCOMI C,. Transcription Factor EB Controls Metabolic Flexibility during Exercise[J]. Cell Metab, 2017, 25(1): 182-196.
[24] NOVAK I, KIRKIN V, MCEWAN D G,. Nix is a selective autophagy receptor for mitochondrial clearance[J]. EMBO Rep, 2010, 11(1): 45-51.
[25] PHILP A, CHEN A, LAN D,. Sirtuin 1 (SIRT1) deacetylase activity is not required for mitochondrial biogenesis or peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1alpha) deacetylation following endurance exercise[J]. J Biol Chem, 2011, 286(35): 30561-30570.
[26] RABINOVITCH R C, SAMBORSKA B, FAUBERT B,. AMPK Maintains Cellular Metabolic Homeostasis through Regulation of Mitochondrial Reactive Oxygen Species[J]. Cell Rep, 2017, 21(1): 1-9.
[27] SAFDAR A, LITTLE J P, STOKL A J,. Exercise increases mitochondrial PGC-1alpha content and promotes nuclear-mitochondrial cross-talk to coordinate mitochondrial biogenesis[J]. J Biol Chem, 2011, 286(12): 10605-10617.
[28] SANCAK Y, BAR-PELED L, ZONCU R,. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids[J]. Cell, 2010, 141(2): 290-303.
[29] SETTEMBRE C, DE CEGLI R, MANSUETO G,. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop[J]. Nat Cell Biol, 2013, 15(6): 647-658.
[30] SETTEMBRE C, DI MALTA C, POLITO V A,. TFEB links autophagy to lysosomal biogenesis[J]. Science, 2011, 332(6036): 1429-1433.
[31] SHAW R J, BARDEESY N, MANNING B D,. The LKB1 tumor suppressor negatively regulates mTOR signaling[J]. Cancer Cell, 2004, 6(1): 91-99.
[32] SMITH B K, MUKAI K, LALLY J S,. AMP-activated protein kinase is required for exercise-induced peroxisome proliferator-activated receptor co-activator 1 translocation to subsarcolemmal mitochondria in skeletal muscle[J]. J Physiol, 2013, 591(6): 1551-1561.
[33] TOYAMA E Q, HERZIG S, COURCHET J,. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress[J]. Science, 2016, 351(6270): 275-281.
[34] WANG L, MASCHER H, PSILANDER N,. Resistance exercise enhances the molecular signaling of mitochondrial biogenesis induced by endurance exercise in human skeletal muscle[J]. J Appl Physiol (1985), 2011, 111(5): 1335-1344.
[35] WU W, TIAN W, HU Z,. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy[J]. EMBO Rep, 2014, 15(5): 566-575.
[36] YAN Z, LIRA V A, GREENE N P. Exercise training-induced regulation of mitochondrial quality[J]. Exerc Sport Sci Rev, 2012, 40(3): 159-164.
[37] YOUNG N P, KAMIREDDY A, VAN N J L,. AMPK governs lineage specification through Tfeb-dependent regulation of lysosomes[J]. Genes Dev, 2016, 30(5): 535-552.
[38] ZHANG C S, LIN S C. AMPK Promotes autophagy by facilitating mitochondrial fission[J]. Cell Metab, 2016, 23(3): 399-401.
[39] ZHAO J, LIU T, JIN S,. Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission[J]. EMBO J, 2011, 30(14): 2762-2778.
The Research Progress of Mechanisms of Exercise-Induced AMPK Regulating Mitochondrial Quality Control
ZHANG Tan1, 2, SUN Yi1, 2, DING Shu-zhe1, 2
1.Key Laboratory of Adolescent Health Assessment and Exercise Intervention, Ministry of Education, East China Normal University, Shanghai 200241, China; 2. East China Normal University, Shanghai 200241 China.
Mitochondria are highly dynamic organelles that under continuous dynamic changes, and the balance between mitochondrial biogenesis, mitochondrial dynamics and mitophagy is crucial to maintain mitochondrial network. The mitochondrial adaption induced by exercise in skeletal muscle includes coordinated improvements in quantity (content) and quality (structure and function). Regular exercise training has known to promote mitochondrial biogenesis, but recent work has demonstrated that it also has a profound impact on mitochondrial dynamics (fusion and fission) and clearance (mitophagy), among which Amp activated protein kinase (AMPK) is considered to play an important role. In this paper, AMPK mediated effect and molecular mechanism of exercise on mitochondrial quality is reviewed.
G804.7
A
1002-9826(2018)06-0097-06
10.16470/j.csst.201806013
2017-01-06;
2018-09-02
国家自然科学基金资助项目(31671241)。
张坦,女,在读博士研究生,主要研究方向为运动适应与线粒体调控, E-mail: zhangtan9999@126.com。
猜你喜欢
世界中医药(2022年18期)2022-11-25
南京医科大学学报(自然科学版)(2022年8期)2022-11-22
重庆理工大学学报(自然科学)(2022年9期)2022-10-26
中风与神经疾病杂志(2022年9期)2022-10-19
中华实用诊断与治疗杂志(2022年1期)2022-08-31
理财周刊(2022年4期)2022-04-30
昆明医科大学学报(2022年2期)2022-03-29
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
昆明医科大学学报(2022年1期)2022-02-28
