
基于词频统计的蛋白质交互关系识别
2019-02-25 13:22蔡松成
计算机技术与发展 2019年2期
蔡松成,牛 耘
(南京航空航天大学 计算机科学与技术学院,江苏 南京 211106)
0 引 言
通过相互作用,细胞中的蛋白质完成细胞中的大部分过程,比如细胞内通讯。因而,蛋白质交互信息(protein-protein interaction,PPI)成为了关键信息,用以解决大量医学难题。目前,生物学家通过人工阅读的方式识别医学文献中的PPI,并按照统一的格式将这些重要的信息录入数据库,如HPRD[1]、IntAc[2]、MINT[3]和BIND[4]等。然而以上数据库中的PPI信息并不全面,而且生物医学的快速发展导致每年相关科学文献的增长数量达上千万,每天也在产生新的蛋白质之间的关系。因此要从医学文献中收集PPI信息,仅靠手工方式难以满足现实的需求。
此背景下,有监督的机器学习方法被大量地运用到研究PPI关系识别中,并取得了巨大进展。由于有监督的机器学习方法需要大量人工标注的数据,代价高昂,所以很多研究者将远监督[5]的思想应用到PPI识别上,以解决训练数据不足的问题。但是由于远监督思想的缺陷,引入了大量的噪音,影响现阶段PPI识别的精度。因此,有研究人员提出了一种基于最大期望算法的蛋白质交互识别方法,构建多实例多标记学习模型,有效消除了签名档中噪音对交互关系识别的影响。文中在该方法的基础上,对每一个蛋白质对的签名档进行词频统计,得到相应的高频词。利用这些信息对最大期望算法的初始化过程进行改进,继而进行PPI识别。
1 相关工作
随着机器学习的流行,研究者们越来越多地采用基于机器学习的方法进行PPI识别。基于机器学习的方法主要包括两大类:基于核函数的方法和基于特征的方法。基于核函数的方法首先对句子结构进行深入研究,通过设计核函数衡量不同蛋白质对间的相似度,然后使用支持核函数的分类器进行PPI关系识别。例如,Bunescu R C等[6]提出了最短依赖路径核;文献[7-9]使用基于多核(特征的核,树核及图核融合)的学习方法抽取PPI信息。基于特征的方法试图从标注有交互关系的句子中抽取重要特征,包括词汇特征、语法特征和语义特征,建立模型来判断蛋白质之间的交互关系[10-13]。但有监督的方法需要大量有标注的数据,且研究对象是单个句子,因此只能依赖一个句子中的线索,对于复杂的句子描述很难判断。
2 基于最大期望算法的PPI识别
文中主要从以下几个方面对基于最大期望算法的多实例多标记学习框架[14-15]加以改进。
2.1 基于高频词的签名档句子筛选
以远监督为基础的关系抽取方法利用已有的知识库和文本集,通过启发式的匹配来提供训练数据。这种方法可以产生大量带标注的训练数据,很好地解决了人工标注数据代价昂贵的问题,节省了人力物力。但是这种匹配是基于假设条件:如果文本集中的语句包含了知识库的实体对,那么这条语句就表达了实体对在知识库中所对应的关系。显然,该假设过于理想化,会产生大量的噪音数据。
作为关系抽取在生物信息学方面的应用,基于远监督的蛋白质交互关系抽取同样面临训练数据存在噪音的问题。首先,利用现有的HPRD数据库查询获取有交互关系的蛋白质对,然后从PubMed数据库中自动获取包含蛋白质对的句子形成签名档。在得到的签名档中,部分句子并未表明目标蛋白质对之间的交互关系。例如,(ahr,rela)是一对有交互关系的蛋白质对,下面是其签名档中的两个句子:
1:We demonstrated that #ahr# associates with #rela# in the cytosol and nucleus of human lung cells.
2:Thus, the #rela# and #ahr# proteins functionally cooperate to bind to NF-kappaB elements and induce c-myc gene expression.
在这两个句子中,第一个句子确实表达了目标蛋白质对(ahr,rela)之间的交互关系,而第二个句子只是包含了目标蛋白质对,但并未表达两者之间的交互关系,所以这个句子成为了该蛋白质对的噪音。训练数据中的这部分噪音会影响最终模型的性能。因此,文中提出了基于高频词的去噪模型来对训练数据中的噪音进行处理。
2.1.1 提取签名档的词频统计信息
通过对蛋白质对签名档数据的观察发现,对于这些描述蛋白质交互作用的句子在单词级别上是存在相似性的。句子中经常出现bind、interact、activate、association、ligand、inhabit、induce等表示蛋白质交互作用的单词。鉴于生物医学文本对于蛋白质交互关系表达的这种规律,试图对每一个蛋白质对的签名档进行词频统计,同时设定阈值,得到对应的高频词集。根据签名档的高频词集,对签名档中的句子进行去噪处理。
提取签名档的词频统计信息主要包括以下步骤:
(1)针对签名档数据,利用NLTK自然语言处理工具包进行词性标注;
(2)两个目标蛋白质之间的单词对描述交互关系更为重要,所以只对这部分单词进行词频统计;
(3)进行单词预处理,删除长度小于2的以及为纯数字的单词,同时将单词中的大写字母转化为小写字母;
(4)进行词频统计时,只考虑名词和动词这两种具有实际意义的单词,这两种词性的单词对蛋白质交互作用的描述起到了非常重要的作用,同时剔除掉名词中的专有名词;
(5)考虑到选择出来的名词中可能包含除目标蛋白质以外的其他蛋白质,因此采用abner蛋白质命名实体工具,识别出其他蛋白质的名称,将其去除掉;
(6)利用NLTK工具包进行英文词干提取。
通过以上6个步骤,最终得到每个蛋白质对签名档下的词频统计信息。
2.1.2 基于高频词的去噪模型
本节根据蛋白质对的高频词集对签名档中的句子进行去噪处理。
算法1:利用高频词集确定句子类别。
输入:
词频阈值TC
词频统计映射表WC
高频词(high frequency words)集合HFW={}//高频词和频率的映射
句子特征词集合F={f1, f2,…,fn}
输出:句子标签集label={r1, r2,…,rm}
1:if m<=C then
2:HFW=WC
3:else then
4:for count in WC. keys() do
5:if count>TC then
6:HFW=HFW∪{count:WC[count]}
7:flag=false //用以判断句子类别的辅助标记
8:for fk∈Fido
9:if fkin HFW. keys() then
10:ri=‘1’
11:flag=true
12:if flag==flase then
13:ri=‘0’
14:return label
算法1描述了如何利用高频词集合确定句子的类别,其中句子特征词集合就是利用2.2.1节提取词频统计信息的6个步骤后,得到每个句子两个目标蛋白质中间的若干一元词。
算法1主要包含两个步骤:
(1)如果签名档的大小小于阈值C,保留词频统计信息作为高频词集;大于阈值C,则对于词频统计映射表中频率大于词频阈值TC的单词才被保存到高频词集中,对应算法1的1~6行,文中设置TC为1。
(2)根据得到的高频词集合来确定句子标签,如果句子的特征词集合中包含高频词,就认为其是有交互关系的,否则认为这个句子不表达交互关系,对应算法的7~13行。
接下来通过获取到的句子标签集合对句子进行去噪处理,算法2描述了基于高频词的句子筛选过程。
算法2:基于高频词的句子去噪处理。
输入:
有交互蛋白质对集P
无交互蛋白质对集N
句子标签集label={r1,r2,…,rm}
签名档中的句子集合S={s1,s2,…,sm}
输出:签名档中的句子集合
1:if
2:for ri∈label do
3:if ri==‘0’ then
4:count_1+=1
5:if count_1==m then
6:randomly save somesentences,others remove from S
7:else then
8:for ri∈label do
9:if ri==‘0’ then
10:removesifrom S
11:else then
12:for ri∈label do
13:if ri==‘1’ then
14:count_2+=1
15:if count_2==m then
16:randomly save some sentences and set the labe1 as ‘0’
17:else then
18:for ri∈label do
19:if ri==‘1’ then
20:remove sifrom S
21:return S
算法2对于有交互的蛋白质对,根据高频词集判断为无交互的句子被认定为噪音数据,要从签名档中去除,对应算法的7~10行。同时,可能一个蛋白质对下的所有句子标记全是无交互的,为了避免把该蛋白质对直接从训练数据中过滤掉,随机保留部分句子作为该蛋白质对的签名档,对应算法的1~6行。对于无交互蛋白质对签名档中的句子,根据先验知识,它们应该都不表达交互关系,但是通过高频词集,同样会把无交互蛋白质对中的部分句子认定为是有交互关系的,这部分句子成为无交互蛋白质对中的噪音,需要去除,对应算法的17~20行。同样,当整个签名档中的句子均判断为有交互时,随机保留部分句子。和有交互蛋白质对不同的是,对于这部分句子,根据先验知识,将其标记改为无交互的,对应算法的12~16行。
2.2 最大期望算法的模型训练
由于蛋白质对签名档中的句子标记是未知的,因此利用最大期望算法在模型存在未知变量的情况下,对模型参数进行极大似然估计。
2.3 最大期望算法的具体实现
在利用最大期望算法实现多实例多标记学习模型的过程中,为了保证模型的学习效果,主要针对最大期望算法的初始化步骤进行了处理。
初始化:由于最大期望算法并不是全局最优算法,无法保证找到全局最优解,因此初始值的选择非常重要。在该模型中,初始值为签名档中句子的类别分布zi,算法起始于M步,在M步利用初始句子类别训练模型中的分类器,然后在E步骤中对初始句子类别重新判断,重复迭代。文中主要设置以下两种初始值:
方案C1:以远监督匹配得到的句子类别作为多实例多标记模型的初始值。既有交互蛋白质对签名档中的句子均为有交互,无交互签名档中的句子均视为无交互。
方案C2:利用基于高频词集合去噪处理后得到的句子及其句子类别作为多实例多标记模型的初始值。
文中采用这两种不同的初始值设置方式试图求得模型的最优解。
3 实 验
3.1 实验数据
实验中有交互关系的蛋白质对是直接从HPRD数据库中查询获取,并且只保留被PubMed数据库中一篇以上摘要包含的那些蛋白质对。而对于无交互关系的蛋白质对,将HPRD中的蛋白质随机组合成蛋白质对,去除已被HPRD数据库包含的蛋白质对以及未被PubMed数据库记载的蛋白质对。以一对蛋白质为查询参数,从文献中检索出描述这两个蛋白质的所有句子,作为该蛋白质对的签名档。最终总共得到有交互关系和无交互关系的蛋白质对分别为576对和578对,合计1 154对。
3.2 实验设置
实验采用的结果性能评价指标是当前PPI抽取系统主要使用的三个指标:精确度(Precision=TP/(TP+FP))、召回率(Recall=TP/(TP+FN))和F值(F-Score=2P×R/(P+R))。为了避免简单应用模型而产生过拟合问题,利用五折交叉验证来评估模型的性能。
3.3 实验结果及分析
为了比较最大期望算法的两种初始值设置方式的实验效果,取最大期望算法迭代的前6次(迭代6次以后实验结果基本趋向局部最优解),对有交互蛋白质对的识别结果如表1、表2所示。
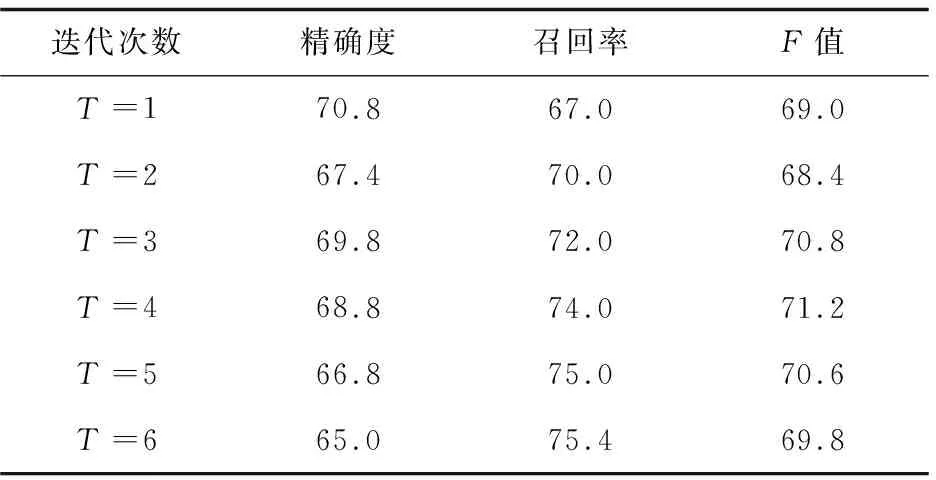
表1 方案C1的识别结果 %
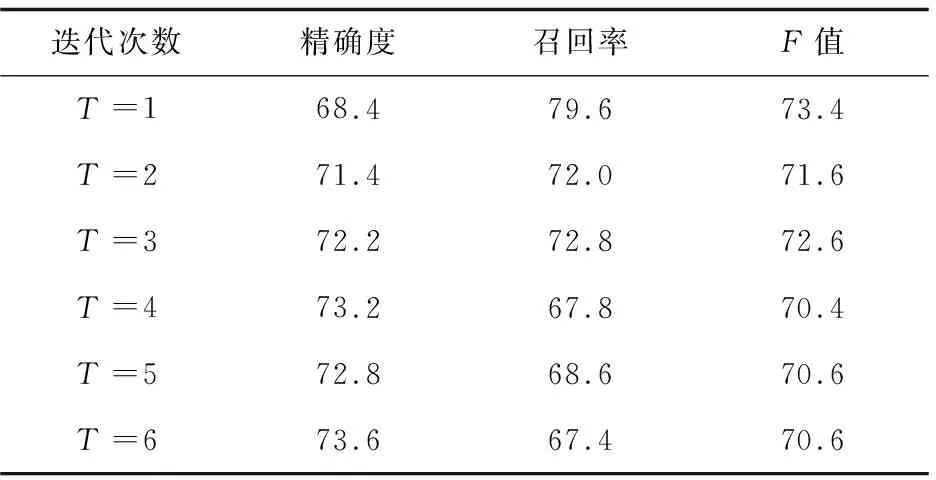
表2 方案C2的识别结果 %
通过对表1、表2的观察可以发现,在前三次迭代,实验方案C2在精确度、召回率和F值上都有明显的提高。随着迭代次数的增加,C2获得了较高的查准率,但是查全率相对较低。说明在基于高频词的去噪模型中,由于存在一部分被判断为噪音数据的句子未被清除(为了避免该蛋白质对签名档的数量为0),导致将一部分有交互的蛋白质对认为是不表达交互关系的,从而获得了较低的查全率,但是整体性能较实验方案C1还是有明显的提高。实验结果说明,不同的初始化方式对本模型的迭代训练过程有很大的影响,同时基于高频词的去噪模型对训练数据的处理是合理的,使模型取得了更好的性能。
4 结束语
在基于最大期望算法的多实例多标记学习框架的基础上,对训练数据中存在的噪音问题进行处理,提出了基于高频词集的去噪模型来改进最大期望算法的初始化步骤,有效提升了蛋白质交互关系抽取模型的整体性能。下一步可以在多实例多标记模型的迭代训练过程中引入一定的先验知识,以进一步提高模型的去噪能力。
猜你喜欢
小康(2022年7期)2022-03-10
小康(2022年7期)2022-03-10
小康(2021年7期)2021-03-15
小康(2021年7期)2021-03-15
意林·全彩Color(2019年9期)2019-11-13
亚太教育(2018年5期)2018-12-01
新城乡(2018年8期)2018-09-03
新城乡(2017年12期)2018-01-04
新城乡(2017年9期)2017-09-26
读者·校园版(2015年7期)2015-05-14
