
美法两国对超说明书用药监管对策及其借鉴启示
2019-03-12 06:01郭俊荣沈阳药科大学工商管理学院辽宁沈阳006解放军总医院药学部临床药学中心北京00853
中国药物应用与监测 2019年1期
郭俊荣,张 方(.沈阳药科大学工商管理学院,辽宁 沈阳 006;2.解放军总医院药学部临床药学中心,北京 00853)
超说明书用药(off-la bel drug use)是药物的使用超出国家药品监督管理部门批准范围,同时也被称为药品说明书外用法以及药品未注册用法[1],主要表现在适应证、剂量、给药途径、给药频次、适用人群、禁忌症等方面,医学的进步和创新与药品说明书更新的滞后是超说明书用药的主要原因。临床上因现有药物无法满足棘手疾病的治疗,医生在循证证据充足、患者知情同意的情况下,往往会超说明书用药。超说明书用药因安全性不可预测,其利益/风险比未知,会给患者造成一定的潜在风险;医生因超说明书用药产生医疗纠纷时往往处于被动地位,带来执业风险;在社会保障方面,超说明书用药相关不良事件产生的费用往往不在报销范围之内,增加患者经济负担。
我国对超说明书用药监管体系比较薄弱,相比来说,美国有最严格的监管体系来保护患者权益,法国在继苯氟雷司事件后也加大了对超说明书用药的监管力度,旨在规范超说明书用药的安全使用。目前我国对超说明书用药医院内部监管对策已有相关报道[2-3],且有些医院内部已有规范的超说明书操作流程[4-5],本文通过对美、法两国超说明书用药的监管对策进行分析,旨为我国超说明书用药监管提供参考。
1 法国对超说明书用药的监管措施
苯氟雷司为降糖药物,主要用于糖尿病的治疗,因其有抑制食欲的功效,后作为减肥药物在肥胖人群中使用,但是超说明书用药引起了致命的心脏瓣膜疾病,该苯氟雷司事件使法国政府对超说明书用药的风险引起了高度的重视。法国政府加大力度制定超说明书用药的监管框架,对超说明书用药采用“临时使用建议(temporary recommendations for use,TRU)”对策[6]。TRU为法国提供了超说明书用药的监管程序,它旨在通过建立超说明书用药患者监测程序和数据收集,规范药品上市许可(marketing authorization,MA)外的使用范围,用于暂时监督未获许可适应证药物的处方。TRU目的是对超说明书使用的药品给予相对较长的观察期,以评估超适应证使用药物的益处和风险,并收集科学严谨的数据以确保超说明书用药的安全性。TRU包括的药品一般用于罕见疾病 或棘手疾病的治疗,常用于肿瘤学、血液学及传染性疾病。
1.1 政府部门对超说明书用药的监管
临床上在面对某些棘手疾病治疗,且没有其他适合的药物可用时,法国药品管理机构(French Medical Regulatory Authority,ANSM)将适应证外药品授权TRU。ANSM由卫生部、社会保障机构、高级卫生局、国家癌症研究所、稀有疾病研究中心等组成。
在超说明书使用药物纳入TRU前需收集有力的科学证据,包括高质量的流行病学研究,但是如果仅有个别案例证据或等级差的研究,则必须开展进一步的临床试验。处于TRU的药品,如果发现对公众健康有潜在风险或缺乏对患者的随访,ANSM可以采取修改、暂停或撤销TRU的决定;如果存在积极的治疗效果,ANSM将评估制药企业和学术研究机构提供的数据,建立患者随访的机制,收集超说明书用药的有效性和安全性数据,并将实际使用数据情况向政府机构部门汇报。
1.2 设立超说明书用药委员会
ANSM虽然采用了一种有效的机制来控制医学上有相对稳定的数据支持合理的超说明书用药,然而这种机制有其局限性,并且将无法控制所有超说明书用药。因此有研究组织[6]建议设立超说明书用药委员会以评估所有超说明书用药情况,并执行以下任务:1)通过咨询和分析现有数据库和查阅国际科学文献来监测超说明书用药处方;2)研究风险人群的群体药代动力学数学模型,如儿童、老年患者、怀孕妇女等;3)实施和监督TRUs的职能;4)在公共健康卫生领域实施监测超说明书用药的机制;5)通过对卫生保健专业人员发送信件、网络出版物、超说明书用药情况等方式向不同健康行为者进行教育计划和交流等。
法国相关研究小组对超说明书用药提出了相关建议[7],具体如下:1)跟踪和监测超说明书用药;2)追踪临床试验中超说明书处方的风险,加强公共研究;3)报告不合理使用超说明书处方,特别是有安全隐患的处方;4)对采用TRU的患者强化监测随访;5)动员所有卫生当局,如法国卫生最高委员会(French National Authority for Health,HAS),ANSM和卫生产品经济委员会(Economic Committee for Health Products,CEPS)等,并确保在卫生部的监管下对超说明书用药进行协调;6)将超说明书用药信息及时告知患者和医疗保健专业人员,使患者有知情权,并提高临床对超说明书用药的风险防范意识。
1.3 建立共享数据库
识别超说明书用药有很多来源,但尚未结构化。超说明书用药处方较难追踪,因此为了识别超说明书用药处方,必须共享数据库。
数据库一般从追踪药物警戒、医院药物监测、制药行业的销售数据库或市场调查等中获取。但是,这些不同资源往往是独立的,尚未共享。科学文献可能会过度强调对公众宣传超说明书用药的做法,此外,媒体上也可能出现一些对超说明书用途的宣传,需进一步核实分 析,以防止超说明书用药信息对大众的误导。
1.4 药品生产企业对超说明书用药的监管
在法国,法律赋予制药公司有监督超说明书用药的责任和义务,制药公司必须监督上市后药物处方的使用情况,并确保药品按照说明书的规定使用,并且不得将未经许可的适应证药品推销给他人。如果观察到非常规处方,制药公司必须立即通知ANSM,并采取一切适当措施通知医疗保健专业人员,以防止超说明书用药的发生。法国政府鼓励制药公司收集超说明书用药的相关可靠证据数据以扩大其上市药品的授权范围。TRU使用期间,制药公司需密切监测患者的随访数据,以收集足够的安全性和有效性数据来扩大MA,同时并为超说明书用药患者的监测提供资金支持,并为患者因超说明书用药产生的损害给予资金补偿。
2 美国对超说明书用药的监管
在美国,医生可以超说明书使用药物,但制药商不能给医生宣传超说明书用药的方法,FDA尽量在为医生提供自由的最佳临床判断和防止制药商不恰当地影响处方决策之间取得平衡。FDA法规旨在确保药品广告和来自临床经验的实践证据真实可靠,无误导性,这些法规适用于期刊、报纸、杂志、广播、电视等的广告。FDA除了对制药公司在互联网上发布的药物信息进行监管,还对直接分发给医疗保健专业人员和患者的材料进行审核。FDA法规规定药品广告不得忽略重要事实或夸大药品疗效,必须在功效、副作用、禁忌症和警告的披露之间达到公正的平衡。
2.1 对制药厂商超说明书促销行为的监管
药品生产企业为了追逐药品带来的巨额利润,常常扩大药品促销[8]。由于药品注册时只批准一种适应证,其他潜在的适应证由于数据有限而未被批准,药品生产企业将一些未被注册的适应证透露给医生和患者,以增加药品使用范围,增加销量[9]。如果允许制药商对药品进行超说明书促销,制药商会尽可能获得最低的药品要求标准,从而用最低标准来影响处方过程,削弱了FDA对处方的安全性和有效性审核评估的力度。
FDA通过直接监督来控制处方药的促销,并得到联邦执法部门的支持。FDA要求制药商提交并审核其宣传材料,制药商的宣传声明必须遵循药品说明书上批准的适应证,并且不得夸大药品功效或低估药品风险,以确保宣传材料准确地总结药物的适应证和风险。FDA可以命令将其认为超说明书的材料从市场撤出,并对制药商当众公开警告。
2.2 超说明书用药参考信息的许可来源
尽管FDA禁止制药公司宣传药品用于未被批准的内容,但医生在超说明书用药时可参考以下信息来源:1)相关资料和药物信息参考:美国的超说明书用药可以参考的权威资料主要有美国医学协会出版的《药物评价》;《美国药典》等。2)期刊文章:药品上市后,制药公司研究人员继续开展临床试验,以开发药物的新用途,将研究数据公开发表于期刊文献。3)医疗联络人:FDA允许制药公司对医师为了寻求证据以支持超说明书治疗做出解答,公司提供的解答必须是非促销性的,并且可能包括期刊文章、处方建议,销售代表可以向医疗保健人员提供同行评审的期刊文章,但不允许使用它们来推广公司产品。4)网站:互联网访问信息的便利性对制药商、处方医师和患者来说是一个优势,美国食品和药物管理局建议应该在网页上显示药物的黑匣子警告信息,并标注处方信息。
2.3 对医师的监管
对医师的处方行为进行监测,应考虑多种因素,包括循证医学证据、是否受药品促销信息的影响以及处方药品可能存在的报销障碍,以避免药物的不合理使用,保护患者免于不安全或无效的超说明书用药,防止临床上医疗事故的发生,确保医生在没有制药商促销的压力下根据自己的科学评估合理地超说明书使用药品。
越来越多的国家和地区正在考虑采用阳光法律[10],该法律要求披露医生和药品生产商之间的财务关系,使医疗专业人员和制造商之间的任何不适当的关系被识别,立法应限制或禁止制药商向医生赠送礼物,医疗销售代表应遵守行为守则,不得参与欺骗性或误导性的营销活动。
医生应了解患者的疾病状态、最佳可用医学循证证据以及药品不良反应等,以为患者选择合适的药物。当通过联邦渠道无法获得超说明书用药信息时,医生可依赖药物专业组织、同行评议的医学期刊和共识指导方针来协助方案的制定。
执业医师在为患者选择超说明书药物时,应提供权威的循证医学证据,确保所选用的超说明书用药方案为最佳使用药物,且其有效性和安全性能得到保障,将风险降至最低。同时,在激烈的医患关系背景下,患者应具有超说明书用药的知情权,应被告知超说明书用药的不确定性,并参与整个复杂的决策过程。
3 对策和建议
超说明书用药应有高质量的循证证据做支持,需经过伦理委员会的批准,并获得患者的知情同意书,建议我国医疗机构在评估超说明书用药使用的适宜性时参考国外文献[11]中图1所示流程,将超说明书用药风险降至最低。针对我国目前超说明书用药监管比较薄弱的情况,借鉴美、法两国监管对策,提出以下建议。
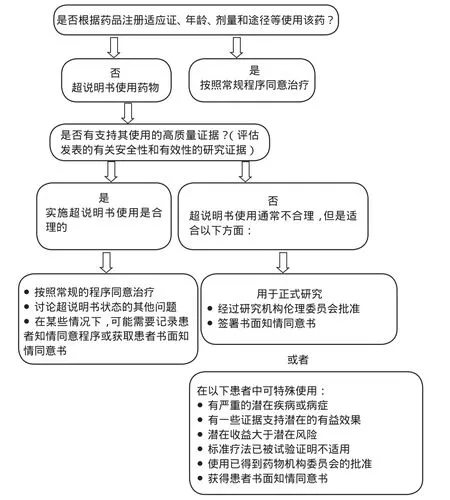
图1 超说明书用药使用的适宜性评估流程图Fig 1 Flow chart of assessing appropriateness of off-label medicine use
3.1 明确超说明书用药的法律地位
由于超说明书用药的存在有其合理性和必然性,但缺乏统一的诊疗规范,应出台相关法律法规,使医生在诊疗过程中做到有法可依,保障医生的权益,提高医生合理使用超说明书用药的积极性,为医学的创新和发展增加动力。
3.2 规范化管理药品说明书
政府部门应该完善和规范药品说明书的审批,确保药品说明书的全面性、科学性、准确性。药品生产企业应对上市后药品进行再评价,收集药品不良反应、超说明书用药的安全性和有效性及不良事件,若某种药品的超说明书用药被广泛认可,政府部门应该开辟快速通道,加强药品注册和药品再评价的联系,鼓励同一药品生产企业联合研究,减少制药企业的资金投入,缩短审批周期,提高制药企业更新说明书的积极性。
3.3 医院内部应规范超说明书用药的操作流程
医院内部各部门要联合起来,药学部门严格审核,药物治疗委员会和伦理审查委员会应严密监督,按照超说明书用药对患者产生危害程度、循证医学证据等级分级管理,明确医生超说明书用药权限,患者知情同意要落实到位,提高医生合理使用超说明书用药的积极性,同时规避执业风险。
3.4 开展循证医学研究
超说明书用药前提是有高质量循证医学证据支持,循证医学工作的有效开展对医学发展功不可没,尤其开展特殊人群的临床试验研究,可为超说明书用药提供重要临床信息。
3.5 明确临床治疗与试验的界限
临床超说明书用药的前提是治疗而非试验,以患者健康考虑,有循证医学证据支持,没有现有药物可选择,患者知情同意,伦理审查要点应涵盖研究的合法性、科学性、严格的试验流程,明确补偿方法和声明利益冲突等。
3.6 加强医务人员药品知识培训,避免超说明书用药风险
临床药师要积极发挥用药指导作用,对药品的安全性、有效性、不良反应等信息能准确地提供给临床,使医生合理恰当选择正确药物,强化超说明书用药的风险意识,保障超说明书用药的规范性。
3.7 加强对药品生产企业的监管
药品生产企业有义务对上市后的药品进行再评价,禁止对医生和患者进行虚假的推销,在医生面对超说明书用药有疑惑时,药品生产企业有义务将前期研究中关于药品的信息告知医生,以提供给医生更多有价值的参考信息。药品生产企业密切监测上市后药品的不良反应,尤其对超说明书用药情况进行跟踪和密切关注,追踪疗效和不良反应发生情况,建议设立基金为超说明书用药患者给予一定的经济补偿,同时要有更新药品说明书的意识。
3.8 加强药品不良反应监测
对超说明书用药患者进行跟踪随访,发现不良反应及时上报,积极干预治疗,将不良反应发生的风险降至最低。
猜你喜欢
金桥(2022年5期)2022-08-24
中学生天地(A版)(2022年6期)2022-07-14
中国药学药品知识仓库(2022年9期)2022-05-23
好日子(2021年8期)2021-11-04
中华养生保健(2021年18期)2021-02-13
循证护理(2021年1期)2021-01-26
中华养生保健(2020年10期)2021-01-18
今日农业(2020年22期)2020-12-14
戏曲研究(2020年1期)2020-09-21
皮肤性病诊疗学杂志(2020年4期)2020-09-02
