
核糖体DNA转录的表观调控与肿瘤发生
2019-03-19 02:44程香荣胡兴琳姜琦黄星卫王楠雷蕾
遗传 2019年3期
程香荣,胡兴琳,姜琦,黄星卫,王楠,雷蕾
核糖体DNA转录的表观调控与肿瘤发生
程香荣,胡兴琳,姜琦,黄星卫,王楠,雷蕾
哈尔滨医科大学组织学与胚胎学教研室,哈尔滨 150081
近年来,表观遗传机制的研究结果提示核糖体DNA (rDNA)表观调控机制的缺陷可能诱导肿瘤发生。ATRX/DAXX复合物通过介导H3.3的H3K9me3修饰,建立和维持rDNA转录沉默。/基因在部分肿瘤中经常发生突变,可能刺激rDNA转录而促进肿瘤发生发展。本文主要阐述rDNA转录表达异常对肿瘤发生的促进作用,介绍rDNA基因转录的表观遗传调控机制,以期为针对rDNA转录调控机制的药物研发提供新的理论支持。
rDNA;表观调控;H3.3;ATRX/DAXX;肿瘤发生
早在19世纪晚期科学家们就发现癌细胞中核仁数量增多和比例增大,20世纪初已将这种大核仁特征列为恶性肿瘤的确诊依据。人非瘤病灶细胞中出现巨大核仁的表现与致瘤风险高度相关。核糖体蛋白S19 (ribosomal protein S19, RPS19)突变的遗传性“核糖体病人”,其早期易患先天性纯红细胞再生障碍性贫血(Diamond-Blackfan anemia, DBA),后期可发展为细胞高度增殖性疾病(癌症),提示核糖体缺陷可能与肿瘤发生有关[1]。因而人们提出核糖体生物合成过程中质和量的改变可能致瘤的假说[2]。目前,核糖体异常如何致瘤是生命科学研究领域的热点方向之一[3]。
核糖体DNA (ribosomal DNA, rDNA)转录为rRNA的过程是核糖体生物合成的限速步骤[4]。基因组中rDNA基因有数百个拷贝,具有重复序列的特征,其转录水平的调控主要有2种方式[5]:(1)通过影响细胞的生长增殖信号通路调节rDNA特异的RNA聚合酶Ⅰ(RNA polymeraseⅠ, PolⅠ)的转录效率;(2)通过表观分子作用机制调节活跃态rDNA拷贝数量的所占比例[6]。前者属于短期调节方式,细胞的营养、生长因子、致癌因素等会上调PolⅠ的转录效率,而基因毒性、代谢压力、饥饿、病毒感染、肿瘤抑制因素等会下调PolⅠ的转录效率;后者属于长期稳定的调节方式,通过建立新的表观遗传修饰状态来调控rDNA的转录潜能,在细胞的生长分化、转化过程中至关重要[6]。本文通过阐述核糖体生物合成异常与癌症发生发展的联系机制,讨论了rDNA的表观调控机制的缺陷可能对肿瘤发生的诱导或促进作用,以期为针对rDNA转录调控机制的药物研发提供新的理论支持。
1 核仁结构与核糖体生物合成过程
核糖体是由4种rRNA分子和约80种不同的核糖体蛋白(ribosomal proteins, RPs)组成的直径为25~30 nm的复合体微粒,负责“中心法则”的mRNA到蛋白质这一翻译过程,与细胞的生长增殖活动息息相关。核仁是细胞核糖体生物合成的重要场所,核仁结构的增大反映出核糖体合成速率的增加。H&E染色(hematoxylin-eosin staining)显示核仁通常表现为单一或多个匀质的球形小体,电子显微镜显示核仁包含3个主要结构:纤维中心、致密纤维组分和颗粒组分[7]。rDNA位于纤维中心,而从rDNA新合成出的rRNA分子主要集中在致密纤维组分,在颗粒组分部位继续加工成熟后,与RPs形成核糖体亚单位,最后被转运至胞质中。核仁的纤维组分包含所有rDNA转录过程所需的物质,包括PolⅠ、上游结合因子(upstream binding factor, UBF)、核仁素和核仁磷酸蛋白等,可以采用银染的方法使其选择性显色。银染核仁组织区域(Ag-stained nucleolar organizing region, AgNOR)的面积大小直接反映出rDNA的转录速率,继而反映出核糖体生物合成的速率[8]。大约400拷贝的rDNA基因根据表观修饰特点和转录功能被分为活跃态、沉默态[9]和中间准备态(poised state)[10],在不同分化状态的细胞中分配着不同的比例。活跃态rDNA在PolⅠ和至少3种基本因子[Rrn3 (TIF-IA)、SL1/TIF-IB (selectivity factor 1, SL1)、UBF]的辅助下转录合成47S pre-rRNA,随后加工形成成熟的18S、5.8S、28S rRNA。另一种rRNA分子5S rRNA在核质中由RNA聚合酶Ⅲ合成,随后被转运到核仁中。各种核糖体蛋白由RNA聚合酶Ⅱ转录,在胞质中翻译成成熟蛋白质后被转运到核仁中。随后28S、5.8S、5S rRNA和49种RPs组装形成核糖体大亚基60S,而18S和33种核糖体蛋白组装形成核糖体小亚基40S,最后,大小亚基都被转运至胞质中构成最终的80S核糖体微粒。在整个过程中,rDNA转录速率是核糖体生物合成过程中的限制性步骤[4]。
2 核糖体生物合成与肿瘤发生的关系
自从发现癌症细胞存在核仁过度肥大且外形不规则的特点后,人们就一直在探索核仁改变与癌症发生之间的因果关系。一个快速增殖过程中的真核细胞每分钟就可产生多达2000个核糖体,而高度增殖的癌细胞更加依赖于核糖体的产生过程。肿瘤细胞为增加其核糖体生物合成的速率,经常突变缺失多个负调控rDNA转录过程的肿瘤抑制基因(和等)[3]。RPs除了作为分子组分和rRNA的分子伴侣参与核糖体生物合成外,还在凋亡、细胞周期停滞、细胞增殖、细胞迁移和侵袭、DNA损伤修复、维持基因组稳定等过程中发挥着重要作用[11]。对于这些“核糖体外功能”(extra-ribosomal function),即核糖体蛋白参与的与核糖体生物合成或总体蛋白质翻译过程无关的其它细胞生理过程[12]),主要是通过p53-MDM2通路介导和调节的,调节机制的异常时也会导致肿瘤发生[11,13,14]。此外,除了p53依赖的信号通路外,RPs还通过c-Myc、E2F-1、ATF4和NF-κB等途径影响到核糖体外功能,这些途径的异常也会诱导肿瘤的发生[11]。本文主要以p53依赖的途径为主,重点阐述核糖体生物合成与肿瘤发生的关系。
2.1 核糖体生物合成与细胞周期
当细胞开始增殖时,蛋白合成的需求就会迅猛增加,以满足细胞分裂时所需的大量结构和功能组分。蛋白质合成速率的增加是通过上调核糖体合成速率来实现的[15],只有G1期达到足够多的核糖体储备时才会允许细胞经过G1-S期限制点[16]。事实上,有丝分裂原和生长因子主要通过以下3种途径刺激细胞增殖过程(图1):(1)通过MAPK/ERK信号通路,激活PolⅠ和RNA聚合酶Ⅲ的转录[6];(2)激活基因,Myc蛋白正向调节核糖体生物合成过程[17];(3)激活mTOR信号通路,诱导促进PolⅠ和RNA聚合酶Ⅲ的转录过程[18]。细胞增殖信号可以激活核糖体的生物合成,同时核糖体生物合成的增加也能促进细胞增殖。在细胞静息状态下,较高水平的p53阻碍pRb的磷酸化过程,使pRb保持低磷酸化水平。低磷酸化的pRb与E2Fs结合,阻止其激活E2F靶基因。E2F的靶基因产物是进入S期所必须的,因此肿瘤抑制蛋白pRb与p53在细胞周期的调控中起了重要作用[19]。在细胞正常生理状态下,核糖体蛋白与rRNA分子结合形成核糖体,留下少量游离的RPs。其中,游离的大亚基核糖体蛋白RPL5、RPL11与5S rRNA形成复合物,通过结合MDM2 (mouse double minute 2, MDM2)而抑制其E3泛素连接酶的功能,剩余未被结合的MDM2降解p53而使其保持稳定水平[20,21]。当抑制或扰乱rDNA转录或成熟过程,如DNA损伤、RP基因突变、药物作用、饥饿状态或致癌基因激活造成核仁压力(核糖体压力)时,多余游离的RPs将更多地结合上MDM2,阻碍P53的蛋白酶降解过程,导致p53水平增高,细胞周期停滞[22,23]。
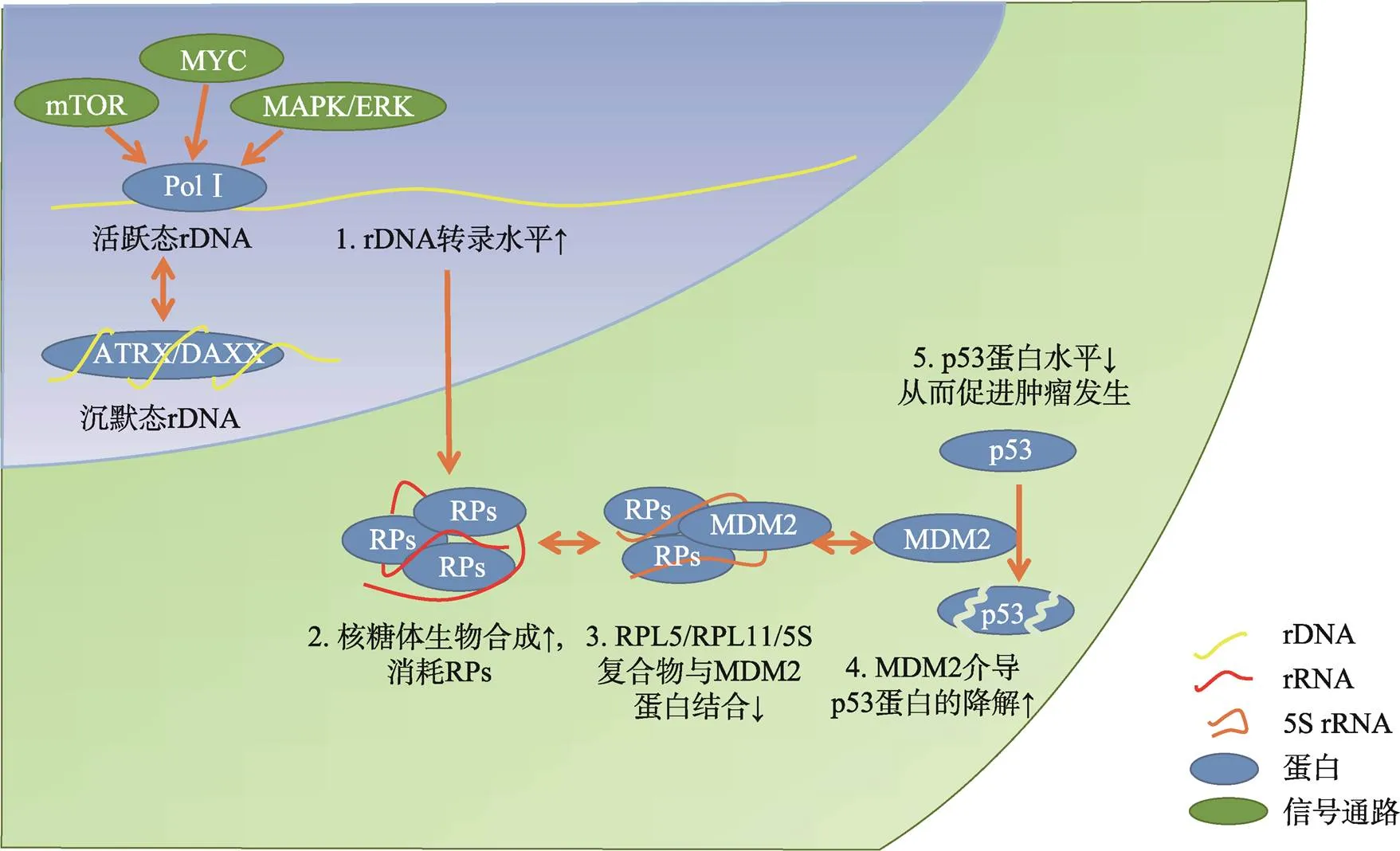
图1 核糖体蛋白-MDM2-p53通路
rDNA转录是细胞活动的中心。mTOR、MYC与MAPK/ERK通路对聚合酶Ⅰ(PolⅠ)的转录具有促进作用,促进核糖体生物合成。核糖体蛋白(RPs)参与核糖体生物合成过程,因而导致游离的核糖体蛋白RPL5、RPL11与5S RNA组成的复合物减少,从而释放出更多的MDM2。游离的MDM2导致p53的降解增多,p53蛋白水平的下降将促进细胞周期进程,从而促进肿瘤发生。本课题组发现ATRX/DAXX复合物与沉默rDNA相关,经常突变基因的肿瘤细胞中,可能是通过开放更多活跃rDNA,从而促进肿瘤发生的重要途径之一。
2.2 核糖体生物合成与肿瘤发生
肿瘤细胞会利用各种方式来提高核糖体的生物合成水平。例如,p53基因的失活是肿瘤细胞中最常见的基因突变方式。p53蛋白负性调节rDNA转录,所以在p53突变缺失的肿瘤中rDNA的转录水平更高[7]。除了p53,肿瘤抑制基因(如和)与致癌基因(如)分别对 PolⅠ转录起着抑制和促进的调控作用[6]。如果抑癌基因和致癌基因发生突变失衡,或在癌症细胞中给予胰岛素或IL-6处理,增强rRNA的转录,就会消耗更多的RPs用于核糖体生物合成,从而减少了与MDM2结合抑制的RPs,导致p53蛋白降解增多,从而游离的p53蛋白水平下降。最终,肿瘤细胞将得益于核糖体合成水平的增高和细胞周期限制的解除,更加快速地增殖和侵袭[20,24]。
核糖体生物合成的“量变”或者“质变”都有可能升高患癌风险。对人类疾病的研究数据表明,患有慢性炎症、二型糖尿病、肥胖的人群中细胞恶性转化的出现频率更高[25~27],这些人具有更高的IL-6或血浆胰岛素水平,IL-6或胰岛素将刺激rDNA的转录过程[20,24]。另外,大量实验研究数据证明了核糖体生物合成在诱导肿瘤发生过程中的重要性。敲除TIP5 (rDNA相关沉默复合物NoRC的大亚基)将导致rDNA的转录水平升高,诱导正常NIH3T3细胞出现转化表型[28]。解除MTG16a蛋白对核糖体DNA的抑制作用,将诱导人正常乳腺上皮细胞出现乳腺癌发生时的形态表型和分子特征[29]。IL-6通过刺激核糖体生物合成过程,诱导人正常结肠黏膜上皮细胞系NCM460出现上皮间质转化、侵袭的特征[24]。因此,核糖体生物合成速率增高的细胞将会有更高的肿瘤发生风险。此外,核糖体生物合成“质变”的“核糖体病人”(如DBA和5q-综合征患者)因其具有核糖体生物合成缺陷而也会具有更高的患癌风险[30,31]。RPs缺陷通常导致细胞低增殖性的表型:如贫血。低增殖特点给细胞带来选择压力,驱使细胞发生二次突变,从而获得高增殖的特性,导致细胞异常克隆并增殖成癌[1,3]。
3 调控rDNA转录的表观遗传异常可能参与肿瘤发生
已分化细胞中大约一半的rDNA基因处于异染色质化的沉默状态[32]。最新研究提示,影响rDNA转录的表观调控机制异常可能是癌症发生的驱动因素,rDNA的表观遗传调控基因突变也可触发肿瘤发生[28,33]。敲减H3K4me3/H3K36me2的去甲基化酶JHDM1B(JmjC domain-containing histone demethylase 1B, JHDM1B)能够诱导rDNA的表观遗传修饰发生重塑,导致转化与未转化的乳腺上皮细胞更具侵袭性[33]。NoRC复合物负责建立和维持rDNA基因的沉默态,敲除其亚基成分将导致沉默rDNA及大小卫星序列的不稳定,rDNA的转录水平增高,从而诱导正常NIH3T3细胞出现转化表型[28]。
外显子测序结果显示,超过60%的胰腺神经内分泌瘤(pancreatic neuroendocrine tumor, PNETs)突变缺失了至少以下3种基因之一:(multiple endocrine neoplasia type 1)、(death domain- associated protein)、(alpha- thalassemia/mental retardation X-linked syndrome protein)[34]。免疫荧光原位杂交实验证实,这些突变与肿瘤的ALT表型(alternative lengthening of telomeres,肿瘤细胞为达到永生而采取的端粒延长机制)相关[34,35]。与胰腺神经内分泌瘤具有相似突变特点的是胶质瘤。例如在儿童多形性胶质母细胞瘤(glioblastoma multiforme, GBM)中,、与(编码组蛋白变体H3.3的基因之一)经常突变,突变频率可高达45%,且与GBM细胞的ALT表型有很强相关性[36]。又如在低级别胶质瘤(low-grade gliomas, LGGs)和继发性胶质母细胞瘤中,突变还经常与(Isocitrate dehydrogenase 1/NADP+)、突变伴随发生[37]。的蛋白产物Menin是组蛋白甲基转移酶复合物的组成成分,参与组蛋白甲基转移酶MLL (mixed lineage leukemia)、PRMT5 (protein arginine methylransferase 5)、SUV39H1 (suppressor of variegation 3-9 homolog protein 1)的功能[38];ATRX是SWI/SNF家族成员的染色质重塑ATP酶,与H3.3特异分子伴侣DAXX组成复合物,将H3.3沉积于异染色质区,建立H3K9me3修饰,从而维持染色质结构稳定[39~41];突变将会促使全基因组范围的DNA和组蛋白的高甲基化状态[42]。考虑到和等基因都具有表观遗传调控的作用,因此该类基因的突变可能重塑了某些肿瘤相关基因的表观遗传修饰状态,有利于肿瘤的发生发展[43]。
虽然突变可以通过诱导重复序列的异常DNA重组而导致ALT表型的出现,使肿瘤细胞永生化[44,45],但ALT并不是肿瘤发生的机制。突变缺失促进肿瘤发生的机制可能是通过诱导基因转录水平改变、DNA修复机制异常来实现的[39]。ATRX综合征患者显示亚端粒区和rDNA基因的DNA甲基化水平降低,特别是rDNA的CpG岛区,这说明ATRX的染色质重塑活性与rDNA的甲基化修饰相关,突变可能对rDNA的表达造成影响[46]。本实验室利用NCBI-GEO数据库中小鼠胚胎干细胞(mouse embryonic stem cell, mESC)的H3.3与H3K9me3的reChIP-seq (连续染色质免疫共沉淀与测序技术)数据[GSE59189][47],采取最近生物信息学分析方法[48]重新分析单个rDNA序列单元上的富集信息。通过分析H3K9甲基转移酶野生型和敲除时H3.3与H3K9me3共富集的情况,结果发现H3.3与H3K9me3主要共富集于rDNA的启动子和编码区,这种共富集状态明显受敲除的影响。本实验室还证明及能够在rDNA的启动子和编码区特异性富集,且随细胞分化过程中沉默rDNA的比例增加而增多。本实验室证明了沉默态rDNA的建立和维持需要ATRX/DAXX复合物与H3.3介导的H3K9me3修饰,敲除将导致rDNA启动子区的甲基化水平降低,UBF结合的活跃rDNA拷贝数量增加等现象(待发表)。在其他报道中,A缺失明显促进胶质母细胞瘤的生长、缩短小鼠的生存期[49]。在胰腺神经内分泌瘤的临床样本中,低表达与更高的(增殖相关的基因)、更高的WHO分级明显相关[50]。以上结果共同提示,或缺失会诱导rDNA转录活性增加,可能是肿瘤发生发展的新机制(图1)。
4 结语与展望
核糖体的快速合成是癌细胞增殖侵袭的基础。核糖体蛋白除了被认为参与核糖体的生物合成外,还具有多种涉及肿瘤发生的核糖体外功能,因此核糖体生物合成过程直接或间接的与肿瘤发生相关。多种肿瘤细胞通过基因突变、代谢方式改变来促进核糖体的生物合成过程,满足细胞代谢需求。ATRX/ DAXX复合物与rDNA的沉默相关,然而其对rDNA转录的表观调控作用是否与ATRX或DAXX经常突变的肿瘤发生直接相关,还需更多的探索。目前,抑制核糖体生物合成已成为抗癌药物研发的重要路径。抑制核糖体生物合成将有利于大量游离的RPs结合MDM2,激活抑癌基因并抑制致癌基因;另外核糖体生物合成不足将阻碍肿瘤细胞的蛋白质合成过程,通过影响细胞周期、细胞凋亡等其他途径,综合抑制肿瘤细胞的生长[51]。抑制核糖体生物合成的药物比起通过引发DNA损伤来激活、诱导细胞凋亡的传统方式,其基因毒性的副作用更小[52]。目前抑制核糖体生物合成的治疗药物主要靶向抑制PolⅠ、翻译起始因子EIF4A、EIF4e、EIF2S1和信号通路mTOR/PI3K,从而抑制核糖体的产生和蛋白翻译的起始阶段[53],未来还可根据rDNA转录的表观遗传调控环节的异常来研发新药。
[1] De Keersmaecker K, Sulima SO, Dinman JD. Ribosomopathies and the paradox of cellular hypo- to hyperproliferation., 2015, 125(9): 1377–1382.
[2] Ruggero D, Pandolfi PP. Does the ribosome translate cancer?, 2003, 3(3): 179–192.
[3] Sulima SO, Hofman IJF, De Keersmaecker K, Dinman JD. How ribosomes translate cancer., 2017, 7(10): 1069–1087.
[4] Kopp K, Gasiorowski JZ, Chen D, Gilmore R, Norton JT, Wang C, Leary DJ, Chan EK, Dean DA, Huang S. Pol I transcription and pre-rRNA processing are coordinated in a transcription-dependent manner in mammalian cells., 2007, 18(2): 394–403.
[5] Cheng XR, Wei YJ, Huang XW, Wang N, Jiang Q, Liu H, Lei L. Research progress on epigenetic regulation of rDNA transcription., 2018,45(5): 485–493.程香荣, 魏艳军, 黄星卫, 王楠, 姜琦, 刘惠, 雷蕾. 表观调控rDNA转录的研究进展. 生物化学与生物物理进展, 2018,45(5): 485–493.
[6] Grummt I. Wisely chosen paths--regulation of rRNA synthesis: delivered on 30 june 2010 at the 35th FEBS congress in gothenburg, sweden., 2010, 277(22): 4626–4639.
[7] Derenzini M, Montanaro L, Trerè D. Ribosome biogenesis and cancer., 2017, 119(3): 190–197.
[8] Derenzini M. The AgNORs., 2000, 31, 117–120.
[9] McStay B, Grummt I. The epigenetics of rRNA genes: from molecular to chromosome biology., 2008, 24, 131–157.
[10] Xie W, Ling T, Zhou Y, Feng W, Zhu Q, Stunnenberg HG, Grummt I, Tao W. The chromatin remodeling complex NuRD establishes the poised state of rRNA genes characterized by bivalent histone modifications and altered nucleosome positions., 2012, 109(21): 8161–8166.
[11] Xu X, Xiong X, Sun Y. The role of ribosomal proteins in the regulation of cell proliferation, tumorigenesis, and genomic integrity., 2016, 59(7): 656– 672.
[12] Warner JR, McIntosh KB. How common are extraribosomal functions of ribosomal proteins?, 2009, 34(1): 3–11.
[13] Zhou X, Liao JM, Liao WJ, Lu H. Scission of the p53-MDM2 Loop by ribosomal proteins., 2012, 3(3–4): 298–310.
[14] de Las Heras-Rubio A, Perucho L, Paciucci R, Vilardell J, LLeonart ME. Ribosomal proteins as novel players in tumorigenesis., 2014, 33(1): 115– 141.
[15] Thomas G. An encore for ribosome biogenesis in the control of cell proliferation., 2000, 2(5): E71–72.
[16] Derenzini M, Montanaro L, Chillà A, Tosti E, Vici M, Barbieri S, Govoni M, Mazzini G, Treré D. Key role of the achievement of an appropriate ribosomal RNA complement for G1-S phase transition in H4-II-E-C3 rat hepatoma cells., 2005, 202(2): 483–491.
[17] Zhu J, Blenis J, Yuan J. Activation of PI3K/Akt and MAPK pathways regulates Myc-mediated transcription by phosphorylating and promoting the degradation of Mad1., 2008, 105(18), 6584–6589.
[18] Mayer C, Grummt I. Ribosome biogenesis and cell growth: mTOR coordinates transcription by all three classes of nuclear RNA polymerases., 2006, 25(48): 6384–6391.
[19] Levine AJ. p53, the cellular gatekeeper for growth and division., 1997, 88(3): 323–331.
[20] Donati G, Bertoni S, Brighenti E, Vici M, Treré D, Volarevic S, Montanaro L, Derenzini M. The balance between rRNA and ribosomal protein synthesis up- and downregulates the tumour suppressor p53 in mammalian cells., 2011, 30(29): 3274–3288.
[21] Donati G, Peddigari S, Mercer CA, Thomas G. 5S ribosomal RNA is an essential component of a nascent ribosomal precursor complex that regulates the Hdm2-p53 checkpoint., 2013, 4(1): 87–98.
[22] Boulon S, Westman BJ, Hutten S, Boisvert FM, Lamond AI. The nucleolus under stress., 2010, 40(2): 216–227.
[23] Russo A, Russo G. Ribosomal proteins control or bypass p53 during nucleolar stress., 2017, 18(1).
[24] Brighenti E, Calabrese C, Liguori G, Giannone FA, Trerè D, Montanaro L, Derenzini M. Interleukin 6 downregulates p53 expression and activity by stimulating ribosome biogenesis: a new pathway connecting inflammation to cancer., 2014, 33(35): 4396–4406.
[25] Roberts DL, Dive C, Renehan AG. Biological mechanisms linking obesity and cancer risk: new perspectives., 2010, 61, 301–316.
[26] Giovannucci E, Harlan DM, Archer MC, Bergenstal RM, Gapstur SM, Habel LA, Pollak M, Regensteiner JG, Yee D. Diabetes and cancer: a consensus report., 2010, 33(7): 1674–1685.
[27] Candido J, Hagemann T. Cancer-related inflammation., 2013, 33, S79–S84.
[28] Guetg C, Lienemann P, Sirri V, Grummt I, Hernandez- Verdun D, Hottiger MO, Fussenegger M, Santoro R. The NoRC complex mediates the heterochromatin formation and stability of silent rRNA genes and centromeric repeats., 2010, 29(13): 2135–2146.
[29] Rossetti S, Hoogeveen AT, Esposito J, Sacchi N. Loss of MTG16a (CBFA2T3), a novel rDNA repressor, leads to increased ribogenesis and disruption of breast acinar morphogenesis., 2010, 14(8), 2186–2186.
[30] Barlow JL, Drynan LF, Trim NL, Erber WN, Warren AJ, McKenzie AN. New insights into 5q- syndrome as a ribosomopathy., 2010, 9(21): 4286–4293.
[31] Doherty L, Sheen MR, Vlachos A, Choesmel V, O'Donohue MF, Clinton C, Schneider HE, Sieff CA, Newburger PE, Ball SE,Niewiadomska E, Matysiak M, Glader B, Arceci RJ, Farrar JE, Atsidaftos E, Lipton JM, Gleizes PE, Gazda HTRibosomal protein genes RPS10 and RPS26 are commonly mutated in Diamond-Blackfan anemia., 2010, 86(2): 222–228.
[32] Grummt I. Different epigenetic layers engage in complex crosstalk to define the epigenetic state of mammalian rRNA genes., 2007, 16 Spec No 1, R21–27.
[33] Galbiati A, Penzo M, Bacalini MG, Onofrillo C, Guerrieri AN, Garagnani P, Franceschi C, Trere D, Montanaro L. Epigenetic up-regulation of ribosome biogenesis and more aggressive phenotype triggered by the lack of the histone demethylase JHDM1B in mammary epithelial cells., 2017, 8(23): 37091–37103.
[34] Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, Schulick RD, Tang LH, Wolfgang CL, Choti MA,Velculescu VE, Diaz LA Jr, Vogelstein B, Kinzler KW, Hruban RH, Papadopoulos NDAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors., 2011, 331(6021): 1199– 1203.
[35] Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C, Bettegowda C, Rodriguez FJ, Eberhart CG, Hebbar S, Offerhaus GJ, McLendon R, Rasheed BA, He Y, Yan H, Bigner DD, Oba-Shinjo SM, Marie SK, Riggins GJ, Kinzler KW, Vogelstein B, Hruban RH, Maitra A, Papadopoulos N, Meeker AKAltered telomeres in tumors with ATRX and DAXX mutations., 2011, 333 (6041): 425.
[36] Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tönjes M, Hovestadt V, Albrecht S, Kool M, Nantel A, Konermann C, Lindroth A, Jäger N, Rausch T, Ryzhova M, Korbel JO, Hielscher T, Hauser P, Garami M, Klekner A, Bognar L, Ebinger M, Schuhmann MU, Scheurlen W, Pekrun A, Frühwald MC, Roggendorf W, Kramm C, Dürken M, Atkinson J, Lepage P, Montpetit A, Zakrzewska M, Zakrzewski K, Liberski PP, Dong Z, Siegel P, Kulozik AE, Zapatka M, Guha A, Malkin D, Felsberg J, Reifenberger G, von Deimling A, Ichimura K, Collins VP, Witt H, Milde T, Witt O, Zhang C, Castelo-Branco P, Lichter P, Faury D, Tabori U, Plass C, Majewski J, Pfister SM, Jabado NDriver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma., 2012, 482(7384): 226–231.
[37] Kannan K, Inagaki A, Silber J, Gorovets D, Zhang J, Kastenhuber ER, Heguy A, Petrini JH, Chan TA, Huse JT. Whole-exome sequencing identifies ATRX mutation as a key molecular determinant in lower-grade glioma., 2012, 3(10): 1194–1203.
[38] Feng Z, Ma J, Hua X. Epigenetic regulation by the menin pathway., 2017, 24(10): T147– T159.
[39] Dyer MA, Qadeer ZA, Valle-Garcia D, Bernstein E. ATRX and DAXX: Mechanisms and Mutations.,2017, 7(3):DOI: 10.1101/cshperspect.a026567.
[40] Goldberg AD, Banaszynski LA, Noh KM, Lewis PW, Elsaesser SJ, Stadler S, Dewell S, Law M, Guo X, Li X, Wen D, Chapgier A, DeKelver RC, Miller JC, Lee YL, Boydston EA, Holmes MC, Gregory PD, Greally JM, Rafii S, Yang C, Scambler PJ, Garrick D, Gibbons RJ, Higgs DR, Cristea IM, Urnov FD, Zheng D, Allis CDDistinct factors control histone variant H3.3 localization at specific genomic regions., 2010, 140(5): 678–691.
[41] Huang XW, Cheng XR, Wang N, Zhang YW, Liao C, Jin LH, Lei L. Histone variant H3.3 and its functions in reprogramming., 2018, 40(3): 186–196.黄星卫, 程香荣, 王楠, 张雨薇, 廖辰, 金连弘, 雷蕾.组蛋白H3变体H3.3及其在细胞重编程中的作用. 遗传, 2018, 40(3): 186–196.
[42] Liu X, Ling ZQ. Role of isocitrate dehydrogenase 1/2 (IDH 1/2) gene mutations in human tumors., 2015, 30(10): 1155–1160.
[43] Elsasser SJ, Allis CD, Lewis PW. Cancer. New epigenetic drivers of cancers., 2011, 331(6021): 1145–1146.
[44] Clynes D, Jelinska C, Xella B, Ayyub H, Scott C, Mitson M, Taylor S, Higgs DR, Gibbons RJ. Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX., 2015, 6, 7538.
[45] He J, Mansouri A, Das S. Alpha thalassemia/mental retardation syndrome X-linked, the alternative lengthening of telomere phenotype, and gliomagenesis: current understandings and future potential., 2017, 7, 322.
[46] Gibbons RJ, McDowell TL, Raman S, O'Rourke DM, Garrick D, Ayyub H, Higgs DR. Mutations in ATRX, encoding a SWI/SNF-like protein, cause diverse changes in the pattern of DNA methylation., 2000, 24(4): 368–371.
[47] Elsässer SJ, Noh KM, Diaz N, Allis CD, Banaszynski LA. Histone H3.3 is required for endogenous retroviral element silencing in embryonic stem cells., 2015, 522(7555): 240–244.
[48] Mars JC, Sabourin-Felix M, Tremblay MG, Moss T. A deconvolution protocol for ChIP-Seq reveals analogous enhancer structures on the mouse and human ribosomal RNA genes., 2018, 8(1): 303–314.
[49] Koschmann C, Calinescu AA, Nunez FJ, Mackay A, Fazal-Salom J, Thomas D, Mendez F, Kamran N, Dzaman M, Mulpuri L, Krasinkiewicz J, Doherty R, Lemons R, Brosnan-Cashman JA, Li Y, Roh S, Zhao L, Appelman H, Ferguson D, Gorbunova V, Meeker A, Jones C, Lowenstein PR, Castro MGATRX loss promotes tumor growth and impairs nonhomologous end joining DNA repair in glioma., 2016, 8(328): 328ra28.
[50] Ueda H, Akiyama Y, Shimada S, Mogushi K, Serizawa M, Matsumura S, Mitsunori Y, Aihara A, Ban D, Ochiai T, Kudo A, Tanabe M, Tanaka STumor suppressor functions of DAXX through histone H3.3/H3K9me3 pathway in pancreatic NETs., 2018, 25(6): 619– 631.
[51] Zhou X, Hao Q, Liao J, Zhang Q, Lu H. Ribosomal protein S14 unties the MDM2-p53 loop upon ribosomal stress., 2013, 32(3): 388–396.
[52] Woods SJ, Hannan KM, Pearson RB, Hannan RD. The nucleolus as a fundamental regulator of the p53 response and a new target for cancer therapy., 2014, 1849(7): 821–829.
[53] Pelletier J, Thomas G, Volarević S. Ribosome biogenesis in cancer: new players and therapeutic avenues., 2018, 18(1): 51–63.
The epigenetic regulation of ribosomal DNA and tumorigenesis
Xiangrong Cheng, Xinglin Hu, Qi Jiang, Xingwei Huang, Nan Wang, Lei Lei
Recent research in epigenetics suggests that defects in epigenetic regulation of ribosomal DNA (rDNA) transcription may contribute to tumorigenesis. ATRX/DAXX complex is involved in the establishment and maintenance of the silence of the rDNA gene through H3K9me3 modification at histone variant H3.3. The ATRX/DAXX-related genes are frequently mutated in some types of tumors, which may increase rDNA transcription and promote cancer development and progression. In this review, we focus on the mechanism that abnormal transcription of rDNA potentially influences tumorigenesis. We also summarize the epigenetic regulatory mechanism of rDNA transcription, which may provide new theoretical support for drug development based on rDNA transcriptional regulation.
rDNA; epigenetic regulation; H3.3; ATRX/DAXX; tumorigenesis
2018-10-15;
2019-01-15
国家自然科学基金项目(编号:31671545) 资助[Supported by the National Natural Science Foundation of China (No. 31671545)]
程香荣,在读硕士研究生,专业方向:基础医学。E-mail: xiangrongcheng@foxmail.com
雷蕾,博士,教授,研究方向:生物发育学。E-mail: leiys2002@yahoo.com
10.16288/j.yczz.18-244
2019/2/25 15:21:42
URI: http://kns.cnki.net/kcms/detail/11.1913.R.20190225.1521.002.html
(责任编委: 宋旭)
猜你喜欢
内蒙古民族大学学报(自然科学版)(2022年2期)2022-11-22
中国畜牧杂志(2020年10期)2020-10-19
理论与创新(2020年9期)2020-07-14
肿瘤防治研究(2020年5期)2020-07-09
生物学教学(2019年9期)2019-09-23
实用肿瘤学杂志(2018年2期)2018-02-01
医学研究杂志(2015年5期)2015-06-10
医学研究杂志(2015年3期)2015-06-10
Zoological Research(2012年6期)2012-08-15
中国病理生理杂志(2010年9期)2010-08-02
