
慢性阻塞性肺疾病的成因及其治疗的困惑与希望
2019-04-27 01:33任成山王关嵩钱桂生
中华肺部疾病杂志(电子版) 2019年2期
任成山 王关嵩 钱桂生
慢性阻塞性肺疾病(chronic obstructive pulmonary disease, COPD)的特征是一种常见的以持续呼吸系统症状和气流受限,呈慢性和进行性发展,通常与有毒颗粒物或有害气体的显著暴露引起的气道和/或肺泡异常相关[1-3]。呼吸系统症状包括呼吸困难、咳嗽和/或咳痰最为常见,其发生率和死亡风险均很高[4]。据世界卫生组织(World Health Organiztion, WHO)估计,全世界约有3.28亿 COPD患者[5],40岁以上的人群患病率为8~10%,因国家和地区的不同,患病率差异很大,最低患病率为2.1%,最高患病率高达26.1%[6],是严重危害人类健康的呼吸系统疾病,是目前全球第四大死亡原因[1-2],到2030年将成为全球第三大死亡原因[7-11]。有研究报道警示,如果吸烟和固体燃料的使用保持现有水平,预测我国到2033年期间将有6 500万患者死于COPD及1 800万死于支气管肺癌[12]。
在过去的20年间,COPD的基础研究和临床实践已经获得了很大的进展,目前已经进行了大量的新药生产并应用于临床。对COPD患者进行个性化和精准化治疗,建立了肺减容术(lung volume reduction surgery, LVRS),其研究成果已证明目前的各种临床治疗方法对COPD患者有一定疗效[13-14]。这些研究结果和临床上取得的疗效推翻了以前认为COPD是一种无望治愈性疾病的论点[15-17]。目前认为,COPD是一种可以预防及治疗的疾病,通过临床积极的治疗,许多COPD患者症状能得到改善,不仅入院率减少,且生活质量和存活率也得到很大提高。尽管COPD已有有效的治疗措施,但是目前还很难阻止或逆转COPD患病后肺功能受损以及加重的趋势,也就是说目前还不能治愈COPD。因COPD发病机制十分复杂,是多种因素、多个环节相互作用的结果,同时COPD伴有重叠综合征、以及COPD伴随疾病多等等[18],所以给COPD的临床治疗带来众多困惑。本文就近年来COPD的成因及其临床治疗的困惑与希望进行论述。
一、COPD成因及发生发展
COPD是一种主要的全球性慢性疾病发病机制十分复杂[19]。但COPD与气道及全身炎症、烟草及环境暴露、合并重叠综合征、伴随疾病及共病、妇女患病率增多、以及COPD罹患支气管肺癌等,并发症及合并症多,给临床治疗带来了极大的困惑。
1. COPD与气道及全身炎症: 气道炎症反应是COPD发病学的关键因素之一,气道炎症是全身炎症的一部分。目前认为COPD是一种复杂的多成份异质性疾病,其特征是慢性全身炎症,且经常与其他伴随多发疾病共存,全身性炎症是COPD患者恶化风险的重要决定因素[20-24]。气道炎症反应在COPD患者肺部中是持续存在的,即使在COPD发病阶段的早中期,也存在炎症及炎症反应[25-26]。急性期炎症反应更趋严重,稳定期炎症反应也不会完全消失。当机体因香烟烟雾、感染及环境暴露污染等因素刺激时,单核细胞(monocytes)、巨噬细胞(macrophages)及多形核白细胞(polymorphonuclear leukocytes, PMN)等迅速合成并释放细胞因子(cytokines),如肿瘤坏死因子-α(tumor necrosis factor, TNF-α)和白细胞介素-8(interleukin-8, IL-8)等。
许多研究表明肺部微生物群与COPD之间存在相关性[27],但是关于COPD患者呼吸道微生物群的变化是否与病情恶化或严重程度相关,一直存在争议[28]。COPD急性加重期并不是特定的肺部微生物群的改变,而是由加重肺部炎症引发的不同生长条件导致的微生物群的失调[29]。急性细菌感染和COPD病情加重之间的比较,突出说明了为什么导致病情频繁发生,以及病情加重的原因是菌群失调所致,并不是由新的细菌感染。全身炎症是气管炎症从肺部“溢出”进入血液循环的结果,而且是COPD涉及多器官及全身的发生[18,30-31]。当然COPD急性感染期间细菌的密度是增高的,虽然在感染期间抗生素治疗有明显的临床益处,但在病情的恶化期间,这种益处是有限的。铜绿假单胞菌(Pseudomonasaeruginosa)病原体引发COPD感染最为常见,且长期可加速病情恶化,致使COPD患者预后较差,有时甚至合并细菌与真菌双重病原微生物感染[32-33]。尽管合理抗生素治疗可能有助于延缓COPD病情恶化,但抗生素可引起肺部或全身环境的变化,最终导致微生物群的失调,甚至持续失调下去[28]。
由于COPD患者长期处于炎症状态,致使小气道纤维化和闭塞,进而导致生理气道通气功能障碍,此种改变比任何肺气肿的发生都为早,小气道纤维化或闭塞的一个潜在机制涉及肺泡上皮细胞的完整性改变[25-26,28]。香烟烟雾和环境空气污染物等诱因可触发这些改变,诱发或加重COPD患者炎症反应,造成细胞因子上调,支气管黏液生成增加,气道通透性增高[34]。总之,COPD气道炎症是一个复杂的临床过程,虽然人们已注意到香烟烟雾与COPD气道炎症存在着密切关系,并且进一步认识到慢性炎症在COPD发生、发展过程中的重要性,见图1[35]。气道炎症无论在COPD急性加重期和缓解期均始终存在,其气道炎症可能在COPD的气道重塑、气道阻塞以肺动脉高压的发生、发展过程中均起着至关重要的作用。继续加强对COPD气道炎症的研究,深入认识气管炎症和全身炎症反应的本质,将有助于研究开发更多新药和新的治疗措施,用于COPD的临床治疗。
2. COPD与烟草及环境暴露: 长期直接或间接暴露于烟草及环境微粒物中是COPD发生发展的主要因素之一,香烟烟雾和环境微粒物中含有超过7 000种化学物质,如氧化气体(oxidative gases)和重金属(heary metals),和至少70种致癌物质[36-37]。就烟草暴露而言,上皮细胞顶端紧密连接决定了上皮渗透性,诱导气道上皮细胞层发生变化,导致杯状细胞增生[38]。并通过选择性自噬途径(autophagy pathway)影响纤毛长度和纤毛循环,以及影响肺泡细胞屏障功能[30,39-40]。此外,香烟烟雾会诱导气道黏膜的通透性,鳞状上皮细胞化生,气道黏液分泌过多,和损伤气道纤毛运动[37]。 由于气道黏膜机制受损, 导致气道黏膜物理屏障活性失活,黏膜纤毛清除系统能力降低,抗氧化剂(antioxidants)、蛋白酶抑制剂(protease inhibitors)和抗菌肽(antimicrobial peptides)生成不足,免疫细胞反应活性下调,这在COPD发病机制中起关键作用[41]。

图1 香烟烟雾初次应答导致COPD;注:TLR:Toll样受体;GM-CSF:粒细胞-巨噬细胞集落刺激因子;HSP:热休克蛋白;ICAM-1:细胞间黏附分子-1;MCP-1:单核细胞趋化蛋白-1;TNF-α:肿瘤坏死因子-α
近年来研究发现,香烟烟雾引起细胞与细胞接触中断,是由于暴露于香烟烟雾中可通过下调α-激酶锚定蛋白-9(A-kinase anchoring protein-9, AKAP-9)的表达,诱导气道上皮细胞E-钙黏蛋白(E-cadherin)介导的屏障功能紊乱[42]。COPD患者肺组织中E-钙黏蛋白表达降低[42-43]。AKAP-9调控蛋白激酶A(protein kinase, PKA)的亚定位(sublocalization),并已与E-钙黏蛋白在基底外侧膜上的定位有关。由于PKA导致环一磷腺苷(cyclic adenosine monophosphate, cAMP)的下调效应因子,并可有助于解释cAMP水平降低导致E-钙黏蛋白表达紊乱的原因。表皮生长因子受体(epidermal growth factor receptor, EGFR)和下调细胞外信号调节激酶(extracellular signal-regulated kinase, ESRK)在气道上皮细胞暴露于香烟烟雾后产生活性氧(reactive oxygen species, ROS)的激活已被观察到[44-45]。香烟烟雾暴露和随后ROS的产生也被证明可诱导EGFR在酪氨酶-845位点磷酸化,导致Src激酶磷酸化并抑制EGFR降解[46]。此外,香烟烟雾已被证明通过相关C3肉毒杆菌毒素(C3 botulinum toxin substrate)1和细胞分裂周期(cell division cycle ccde)42以及P120连环蛋白依赖机制(cafenin-dependent)诱导EGFR活化[47-48],见图2。
环境中的生物质烟雾是一种污染物,这些物质可以用燃料。最常见的是木材、农业残留物,如树枝和干草、动物粪便和木炭[49]。这些物质一般在通风不良的壁炉和炉具中燃烧,产生大量有害污染物。燃烧物质产生的烟雾含有超过250种有机化合物,可产生多种气体污染物,包括一氧化碳(carbon monoxide)、氨(ammonia)、氢氰酸(hydrocyanic acid)、甲醛(formaldehyde)、氮氧化物(nihogen oxides)和硫(sulfur),以及挥发性有机化合物(volatile organic compounds),如苯(benzene)和多环芳烃(polycyclic aromatic hydrocarbons, PAH),如苯并芘(benzopyrene),后两者是人类的强致癌物质[50-51]。
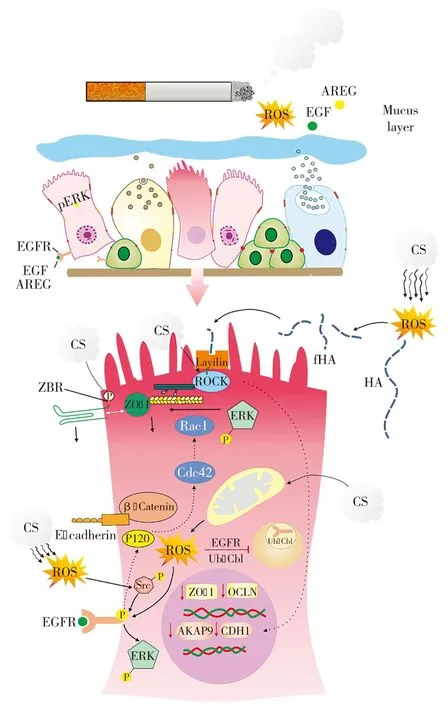
图2 暴露于香烟烟雾后气道上皮连接的破坏;注:在香烟暴露在烟雾中,气道上皮细胞,特别是细胞间接触发生了显著变化。香烟烟雾可导致TJs的破坏,在磷酸化的ZO-1结合酪氨酸OCLN中的ZBR残留。此外,它还能降低ZO-1和OCLN的基因表达。香烟烟雾也能增加线粒体活性氧(ROS)的产生,而ROS反过来又会被激活表皮生长因子受体(EGFR)通过Src介导的ERK信号磷酸化。激活ERK可诱导TJ离解。此外,香烟烟雾还会破坏细胞与细胞之间的接触通过EGF和双调节蛋白(AREG)配体激活EGFR依赖的机制,以及EGFR独立的机制。此外,存在于香烟烟雾中的ROS可以诱导透明质酸裂解,通过其表面受体层林导致Rho激酶(岩石)磷酸化。岩石活化可以通过降低E-钙黏蛋白基因和蛋白表达来破坏AJs。AKAP:激酶锚定蛋白;CDH1:钙黏蛋白;CS:香烟;fHA:支离破碎的透明质酸;HI:透明质酸;Ub-Cbl:E3泛素连接酶。
全球约有30亿人暴露于生物质烟雾中,而吸烟者仅为10亿人。研究表明,生物质烟雾比烟草烟雾更容易导致COPD的风险因素[49,51]。由于社会文化的原因,妇女和儿童是接触这种污染物的主要人群,全球每年200万妇女和儿童死亡[52]。在这些人群中平均要花费6万多小时在生物炉子上做饭,在此期间她们会吸入超过2 500万升的污染空气[53]。生物质烟雾除含有有毒气体外,还含有固体和液体颗粒的混合物,这些颗粒的浓度、大小、表面和化学成分各不相同,称为颗粒物(particulate matter, PM)[50]。有些粒子是可以经呼吸进入肺内,并根据其动力学直径进行分类,PM10颗粒的空气动力学直径为10 μm或更小,PM2.5颗粒的空气动力学直径为2.5 μm或更小,PM0.1,超细颗粒的空气动力学直径为0.1 μm或更小。PM10颗粒通常由含有晶体或非晶态成分的矿物组成,以及从细菌(bacteria)、真菌(fungi)或内毒素(endotoxins)等多种来源吸收的物质[51,54]。PM2.5颗粒金属含量高,一般由碳核组成,其表面吸附有机和无机成分,增强了其氧化应激产生的潜力[55]。PM0.1超细颗粒中多环芳烃含量高,也是强氧化应激的诱导因子,吸入这些污染物对健康是有害的,并且是COPD的高危危险因素[55-59]。
3. COPD与合并重叠综合征: 重叠综合征(overlap syndromes, OS)是一种临床常见的情况,即临床和/或实验的两个或两个以上独立的疾病实体存在于同一患者,在临床上不但增加了准确诊断的复杂性,而且影响了治疗的选择[33]。狭义的COPD与OS只限于COPD和睡眠呼吸暂停低通气综合征(sleep apnea hypopnea syndrome, SAHS)[60]。目前认为,COPD与OS包括支气管扩张(bronchiectasis-COPD overlap syndrome, BCOS)、纤维化(fibrosis-COPD overlap syndrome, FCOS)、哮喘(asthma-COPD overlap syndrome, ACOS)和阻塞性睡眠呼吸暂停(obstructive sleep apnea-COPD overlap syndrome, OCOS)[33]。因此,有必要探讨COPD与合并OS的危险因素、病理生理、诊断和潜在治疗的差异,将COPD与OS作为不同的疾病状态进行识别和区分是至关重要的。同时注意关注呼吸OS,即COPD之间的重叠,哮喘,支气管扩张和COCS。大多数呼吸道COCS的核心是COPD,慢性炎性疾病的特点是持续的气流阻塞,加速肺功能丧失和不可逆的肺损伤,见图3[33]。在全球范围内COPD患病率和病死率的增加和WHO估计,全球所有死亡者的5%可归因于COPD[61]。虽然COPD的主要诱因通常是暴露在烟草烟雾中,但其他日益增加的危险因素,包括空气污染和职业暴露,重要的是与哮喘、支气管扩张和COCS重叠[62]。
哮喘和COPD是国际上最常见的呼吸系统疾病[63-64]。虽然两者都有共同的特点,如气道炎症和气流受限,大多数学者认为两者是不同的疾病状态,其病因、病理生理、预后和治疗反应各不相同,见图3[65]。但情况并非总是如此,早在20世纪60年代,有学者提出,所有的气道疾病,包括哮喘、肺气肿和慢性支气管炎,都应该被认为具有共同遗传起源的“慢性非特异性肺病”(chronic nonspecific lung disease)[33,66-67]。但也有学者持不同观点,认为哮喘和COPD是两种截然不同的疾病,其因果机制也不尽相同,这些争论持续了半个多世纪,在某种程度,不同观点各有其价值[33,68-69]。ACOS是对这两种疾病的临床特征同时存在的一种认识[63-64],全球哮喘防治创议(Global Inifitative for Asthmea, GINA)和全球COPD防治创议(Global Inifiative for Chronic Obstructive Lung Disease, GOLD)指南都承认这一点,并建议临床在评估哮喘和COPD的特征时,要考虑ACOS的诊断。因此,ACOS的早期评估,要考虑到COPD人群的哮喘症状,然而,考虑评估哮喘人群的COPD也很重要,不过后者不甚常见[70-74]。由于缺乏标准化定义,ACOS的真正流行程度存在很大差异,而标准化定义是最近才提出来[74-75]。ACOS文献报道的病死率也有很大的差异[76-79]。
支气管扩张是一种不可逆的结构性气道扩张,由高分辨率计算机断层扫描(high-resolution computed tomography, HRCH)识别,这与COPD形成对比,COPD的生理学诊断是基于不可逆的气流阻塞、症状的严重程度和加重的频率[63,80-81]。肺纤维化(pulmonary fibrosis)是一种导致瘢痕形成的肺间质疾病。肺纤维化和COPD称为FCOS,其临床特征得到了新兴证据的支持,这些证据将两种疾病的共同特征与共同的病理生理学基础联系起来[82-83]。OSA是由于上气道的间歇性塌陷,从而导致吸气气流减少或缺失,其特征是阻塞性睡眠呼吸事件,表现为呼吸暂停、低通气和苏醒,睡眠中断的迹象和睡眠质量差的日间症状[84]。学者们认识到微生物感染在COPD与OS发生发展的作用,为开发新的诊断和治疗方法奠定了基础[85]。其中微生物组的组成及其干扰,在呼吸道感染中显然发挥着重要作用[86-88],这对于COPD与OS的检测和管理具有潜在的临床意义。微生物组分析显示,肺部以假单胞菌(Pseudomonas)、链球菌(Streptococcus)、普雷沃特氏菌(Prevostella)、梭菌属(Fusobacterium)、嗜血杆菌(Haemophilus)、韦氏菌(Veillonella)和卟啉单胞菌属(Porphyromonas)等菌属为主[89]。微生物组中的成分、细菌载量的变化或特定病原体种类的存在可能伴随特定的慢性肺部疾病[88,90-92]。

图3 COPD与目前描述的包括哮喘在内的主要呼吸重叠综合征的中心地位的非比例维恩图,支气管扩张,纤维化和阻塞性睡眠呼吸暂停;注:慢性阻塞性肺疾病(COPD),阻塞性睡眠呼吸暂停(OSA/OSAS),哮喘-COPD重叠综合征、BCOS-支气管扩张症-COPD重叠综合征、FCOS-纤维化-COPD重叠综合征和OCOS-OSA-COPD重叠综合征,HRCT-高分辨率计算机断层扫描;ACOS:哮喘-COPD重叠综合征
4. COPD与伴随疾病及共病: 伴随疾病又称共病(comorbidity),临床上也称多重病症(multimorbidity)。有关共病的英文表达形式主要有3种:comorbidity、multimoorbidity、multiple chronic conditions(MCC),其内涵和外延均存在一定差异。共病(comorbidity)强调“共因”,而多重病症(multimorbidity)仅强调“共存”。comorbidity一词最早由美国学者于1970年提出,是指患有所研究的某种索引疾病的患者同时还伴发其他的疾病[93]。也就是说只有同一患者存在两种明确的疾病时才会被视为共病,或者说是因果关联,有共同的危险因素和病因,并不是说把某些综合征、不良状态等纳入为共病的范畴。comorbidity一词同时发展的还有multimorbidity,由德国学者于1976年提出[94]。随着对其的研究不断深入,2008年WHO将multimorbidity正式定义为同一患者体内存在两种或两种以上的慢性病[95]。这一概念明确地将研究关注点从所界定的索引疾病转为同时患有多种疾病的患者本身。
COPD是一种主要的全球性慢性疾病,而COPD患者普遍存在有共病[19,21,31,96]。一项对213例COPD患者的观察研究中,几乎所有的COPD患者都发现有一种或多种共病(97.7%),54.0%的COPD患者至少有四种共病[97]。COPD患者还伴有合并症,这些合并症往往更频繁地聚集在一起[97]。COPD最常见的并发症包括心血管疾病(cardiovascular diseases, CVDs)、代谢紊乱(metabolic disorders)、骨质疏松(osteoporosis)、骨骼肌功能障碍(skeletal muscle dysfunction)、焦虑/抑郁(anxiet/depression)、认知障碍(cognitive impairment)、胃肠道疾病(gastrointestinal diseases)和呼吸系统疾病(respiratory conditions),如哮喘(asthma)、支气管扩张(bronchiectasis)、肺纤维化(pulmonary fibrosis)和支气管肺癌(bronchogenic carcinoma)[20,98]。焦虑和/或抑郁并在COPD患者更容易加重病情、频繁住院、生活质量下降和病死率升高[18,99]。
尽管COPD及其共病之间的发生机制尚未完全明了,但已经提出了一些潜在的决定因素[21,98,100-101]。老化与多种慢性疾病的发生密切相关,衰老的特征是慢性、迟缓的全身炎症反应,这是大多数与年龄有关的疾病的共同因素,包括COPD遗传易感性是另一个被描述为潜在联系的内在因素[21,102]。有学者报道了男性COPD患者血管紧张转换酶基因(angiotensin converfing enzyme gene)DD基因型与右心室肥厚的发生负相关。同样一些候选基因可能解释肺癌和COPD之间的关系,超过了吸烟作为一种共同风险因素[18,103]。已知COPD和肺癌相关的候选基因(FAM13A、HHIP、ADAM19和CHRNA)与参与气道炎症和凋亡的气管上皮受体相关[104]。目前认为COPD与其共病相关的其他可能机制包括吸烟和缺乏体育活动等常见的危害因素以及氧化应激和全身炎症等常见途径[21,101]。由于全身炎症在COPD及其主要共病的发病机制中所起的作用,致使循环中升高2到4倍的促炎和抗炎细胞因子,如IL-β1和IL-6和TNF-α,所产生的细胞因子拮抗剂和急性期反应蛋白(acute phase proteins),如C-反应蛋白(c-reactive protein, CRP)、血清淀粉样蛋白A(serum amyloid A)[105]。伴随COPD的许多共病以持续的全身炎症为特征[31]。
5. COPD与妇女患病率增多: COPD不再是主要影响男性的呼吸系统疾病,近年来女性COPD患病率不断攀升,在美国COPD是女性吸烟者的主要死因,排在肺癌和CVDS之前[106-107]。重要的是医学界需要改变对COPD仅作为一种男性疾病的概念,目前认为女性COPD的患病率与男性相当[108]。虽然女性吸烟趋势滞后了近一个世纪,但香烟很快在女性中普及,女性吸烟的趋势随着女性地位的变化而上升,似乎与男性吸烟趋势遵循同样的模式,这一趋势与吸烟人数的急剧增加是密切相关的。对于女性来说,吸烟始于两次世纪大战之间,她们渴望解放。通过吸烟,象征着女性是一个享有特权的社会团体成员。从上世纪50年代到70年代,香烟渗透到女性社会的各个阶层,在女性中并变得司空见惯(commonplace)[108-109]。这种现象得到了广告的帮助,将香烟描绘成一种诱惑的手段,先是赞美烟草的减肥效果,然后拍摄一些迷人的女演员[108,110]。然而,一个令人鼓舞的趋势正在西方出现,在美国,女性吸烟被视为负面的,其有害健康的后果是公认的[108]。女性吸烟人数的增加不一定是不可避免的,20世纪中国女性吸烟率下降。本世纪初,女性吸烟率为25%,在20世纪30年代进一步上升,但在20世纪下半叶有所下降。这一逆转与反对妇女吸烟的新的社会文化规范的出现有关。这些新规范是通过传统价值观的运动来传达的,这种价值观的推崇有利于促进身心健康的行为[108]。
从女性COPD的流行病学分析,去医院就诊的女性吸烟者被诊断为COPD的可能性比男性吸烟者低三分之一以上[111]。据文献报道,女性在COPD的诊断上存在延迟,原因是COPD自愿推迟就诊,由于疲劳或抑郁症的普遍存在,导致了一种临床上治疗的差异[112]。目前估计不同的国家和地区COPD的患病率在4.5%到10.2%之间[113-114]。2008年,WHO估计全球有1.68亿男性和1.60亿女性吸烟者[115]。WHO的一份报告还指出,有证据表明烟草广告越来越以年轻女孩为目标,强调吸烟的妇女在社会上的可接受性,并强调吸烟是妇女的解放标志。女性和男性的吸烟率与女性赋权之间存在密切的联系,这种相关性显然与经济发展水平相关[116]。增强妇女权力的趋势是否也会导致烟草在妇女中流行?为强调全球妇女烟草使用增加所带来的风险重要性,WHO 2010年组织的世界无烟日的主题是“性别与烟草”,重点是向妇女推销。但是,在发展中国家的妇女由于燃烧生物质燃料而暴露在室内烟雾中[117-120]。致使女性更容易受到烟草暴露的影响,虽然香烟消费量较低,但COPD发病较早,FEV1下降较快,但相反,停止接触烟草可获得更大益处[121-122]。
女性COPD患者的临床表现似乎与男性不尽相同,女性表现出更多的症状,如呼吸困难或咳嗽[123-124]。即使她们的痰液比男性少,但她们的气道壁更厚[125]。从COPD遗传流行病学分析,显示女性吸烟者COPD早发严重,且风险更高[126-127]。可能与X染色体有关,因为母亲患COPD增加了吸烟的女儿患COPD的风险[127]。解剖因素和发育不良的概念可以解释为什么女性患气道疾病的风险增加,在青春期,女性气道的生长相对于男性的肺生长较小,男性的支气管肺生长较为均匀,因此吸入的物质对较小的表面的影响较小[108,128]。健康女性气道颗粒沉积大于男性,尤其是近端气道颗粒沉积[108]。其次是女性吸烟者的呼吸道局部炎症反应比男性吸烟者更为强烈,呼吸性细支气管炎可能发生在年轻女性吸烟者早期[129]。肺气肿患者气道狭窄与支气管壁增厚增高有关,COPD男性和女性吸烟者肺泡巨噬细胞中某些蛋白的表达也存在显著差异[123,130]。女性COPD患者溶酶体功能受损会导致肺泡巨噬细胞自噬抑制,从而刺激某些细胞因子的产生,导致血清水平升高,这被认为与女性COPD患病率增加有关,但与男性无关[131]。在气道梗阻患者中,女性COPD患者血清IL-16和VEGF水平明显高于男性患者[132]。支气管高反应性(bronchial hyperresponsiveness, BHR)女性比男性更为常见,以及过度吸烟(>20 g·d-1)女性比男性发生重大风险更高[108]。BHR是男女吸烟者肺功能加速下降的危险因素,但女性FEV1下降速度更快,吸入烟雾的方式可改变BHR水平,女性倾向于深吸气,从而加重BHR。未接受激素替代治疗的绝经后女性气管阻塞程度高于接受激素替代治疗的妇女,因为吸烟具有潜在的抗雌激素作用,可能导致女性吸烟者肺功能受损。吸入烟雾后,化学物质影响细胞色素P450酶诱导剂介导,增加了气道中的氧化应激,使女性在吸烟时肺部更容易受到氧化损伤[108,133]。
6. COPD与罹患支气管肺癌: COPD似乎增加了罹患肺癌的风险,而且COPD和肺癌具有共同的临床特征,两者具有较高的患病率和共同的危险因素,如吸烟、某些遗传背景、环境暴露和潜在的慢性炎症反应[134-135]。COPD因其较高的患病率和病死率而导致肺癌预后恶化[136]。所以,肺癌是世界上第一位癌症死亡原因,占所有癌症死亡的13%,每年全球肺癌患者死亡超过1 400 000例[137-138]。烟草消费是肺癌的主要危险因素,因为超过85%的肺癌病例发生在目前或以前的吸烟患者中。烟草也是COPD发生的主要原因。有学者分析了COPD患者并发肺癌的风险,发现肺癌发病率为16.7/1 000人/年[139]。有较重的吸烟史患者患肺癌的风险甚至更高,96.6%的COPD肺癌患者为吸烟者,吸烟量越大患肺癌的风险越高[139-144]。第三军医大学新桥医院2008年1月至2012年12月5年间收治COPD合并肺癌患者122例,有吸烟史者107例(91.0%),吸烟指数为(910.0±457.7/年支),单纯肺癌组吸烟指数为(764.7±383.3/年支),两者比较有统计学意义(P<0.05)[144]。虽然有些COPD和/或肺癌患者从不吸烟,但是吸烟是导致COPD和/或肺癌的主要因素之一,我们曾提出设想:吸烟→COPD→肺癌发生发展三部曲之规律[145]。
长期以来,学者们一直认为慢性炎症与癌症之间存在关联[146]。似乎涉及多种免疫细胞的复杂炎症过程会导致组织损伤和重构,从而导致COPD和隐秘肺癌的发生[147-148]。在过去的10年中,人们对生物标志物的识别产生了极大的兴趣,至少在COPD发生发展中是这样的[149-150]。一些研究甚至观察到吸入皮质类固醇的患者降低了肺癌的风险[151]。如果COPD伴随着致癌烟草化合物的作用而出现慢性炎症通路,那么由COPD引起肺癌的几率可能会增加[145-146]。正因为如此,在被诊断为COPD或肺癌的患者中,吸烟暴露的比例很高[146]。因此,COPD和肺癌两种疾病共存也可以反映共同的吸烟暴露。有学者提出包括肺癌患者在内的研究中发现COPD患病率为50%,基于这些结果,发现20%的吸烟者可能会得COPD,10%的吸烟者可能会得肺癌,如果50%的吸烟者已经患有COPD,那么25%的COPD吸烟者可能会得肺癌[152]。很显然,吸烟在COPD以及肺癌发生发展中的有重要作用,事实上,大约85%的肺癌发生在吸烟者,而95%的同时出现COPD的肺癌患者同样是吸烟者[140,146]。
虽然COPD和COPD并发肺癌的共同发病机制非常复杂,但主要是烟草暴露、慢性炎症、炎症和炎症介质、炎症微环境(inflammatory microenvironment)、免疫紊乱(immune dysfunction)、细胞凋亡(cell apoptosis)、细胞外基质退化、血管再生受限、DNA被破坏、核转录因子-κB(nuclear transcription factor-κB, NF-κB)、α1-抗胰蛋白酶(α1-antitrypsin, α1-AT)缺乏、氧化应激(oxidative stress)等因素[145]。近年来研究发现驱动突变致癌基因在鳞状细胞癌(squamous cell carcinoma)中被修饰,包括成纤维细胞生长因子Ⅰ型受体(fibroblast growth factor receptor Ⅰ, FGFRⅠ)、磷酸酶基因10(phosphatase and tensin homolog 10, PTEH10)以及CDRN2A基因(P16)。学者们证实了鳞状细胞癌组织中的FGFR,会被放大而发生恶变[153]。由于这些潜在强大的驱动突变致病基因还没有新的抑制措施;因此,必需进一步开发治疗肺癌的分子靶向药物(molecular targed drug)。炎症微环境下启动STAT3与NF-κB的致癌作用[154],见图4。
二、COPD治疗困惑与希望
COPD的成因及发生发展非常复杂,主要是气道及全身慢性炎症和炎症反应,吸烟以及环境暴露。人们普遍认为COPD与异常的先天性免疫反应有关,被称为“先天性免疫性疾病(archetypical disease of innatr immunity)”因为吸入颗粒物和有害气体会引发炎症反应[155],导致小气道组织增殖和肺实质组织破坏,免疫细胞还会聚焦到这些小隔间中[156]。所以,吸烟作为COPD的主要危险因素的模式正开始转向接触生物质烟雾(biomass srnoke, BS)暴露,因为全世界每天约有30亿人接触BS污染物[157]。气道及全身炎症不易驱除,先天性免疫不足和BS暴露无法避免。

图4 炎症微环境下启动STAT3与NF-κB的致癌作用;注:NF-κB: 核转录因子;IL-6:白细胞介素-6;COX-M: 环氧化酶;EGFR: 表皮生长因子受体;IGFR: 胰岛素样生长因子受体;VEGF: 血管内皮生长因子;HIF: 缺氧诱导因子STAT3: 信号转导与转录激活因子3;COPD:慢性阻塞性肺疾病
COPD及其重叠综合征、伴随疾病或共病多,这是老年人逐渐发生趋势,一旦发生改变,特别是COPD女性患病率增多,以及COPD罹患支气管肺癌,这些均给COPD患者的治疗甚至治愈带来了极大的挑战,使之对COPD的治疗非常困惑。
尽管目前还没有任何一种药物可以逆转COPD病情进展或者有效地阻止肺功能的恶化。但现在还是已有多种治疗方法,突出的是个体治疗水平上仍然需要进一步优化治疗策略。着手恢复COPD气道上皮屏障活性的最新治疗方法,气道重塑是COPD的一个标志,针对上皮屏障功能恢复的治疗干预措施可能对COPD是有益的[37]。目前COPD的治疗旨在抑制炎症和支气管扩张,包括吸入皮质类固醇(inhaled corticosteroids, ICS)和长效支气管扩张剂(long-acting bronchodilators, LABA)。这些药物可以缓解COPD的发展,并在病情恶化期间暂时缓解症状[158]。有研究显示,COPD患者在接受ICS治疗后肺功能得到改善[159]。利用基因富集和全基因组分析表明,肺功能的改善与COPD上皮屏障功能富集基因的上调有关[160]。表明ICS可能影响COPD上皮屏障功能,并进一步支持屏障功能丧失与COPD肺功能下降相关。除了ICS,还有学者研究表明,在体外用cAMP-evevating复合治疗可以成功地恢复香烟烟雾提取物或转化生长因子(transforming growth factor-β, TGF-β)诱导的气道上皮屏障功能障碍[43,161-162]。因此,PDE4抑制剂对COPD上皮屏障功能障碍的影响具有重要意义[37]。
有研究表明,外源性TGF-β1通过上调紧密连接蛋白(zonula occludens, ZO-1)和ZO-2恢复香烟烟雾提取物诱导的气道上皮屏障损伤。这与之前TGF-β1在COPD组织重塑中的作用形成对比[163]。EGFR预处理气道上皮细胞在体外也被证明可以保护上皮紧密连接不受香烟烟雾提取物诱导的连接损伤,并在体外促进气道上皮细胞修复,这与EGFR在烟雾诱导的屏障功能中的作用再次形成对比[164-165]。关于药物抑制在恢复上皮紧密连接活性的有效性,有人对呼吸道合胞体病毒诱导的气道屏障破坏机制有了新的认识,并表明PKD抑制可减弱呼吸道合胞体病毒诱导的体外连接被破坏[166]。抑制PKD3已被证明可以增强体外气道上皮细胞功能,进而上调CLDN1[167]。使用AKAP抑制剂st-Ht31肽被证明可以抵消香烟烟雾提取物对16HBE细胞中E-钙黏蛋白引起的细胞接触的损伤,因此可能具有治疗效果[42]。通过恢复上皮细胞屏障功能,调节EMT和上皮细胞修复的药物干预,特别是针对EGFR及其下调信号的药物,以期恢复COPD患者气道屏障功能可能是有益的。
基础实验研究评价治疗COPD的疗效是一回事,临床实际治疗COPD患者,判断观察疗效是另一回事,就目前而言,COPD治疗的目的是减轻症状,减少加重的频率和严重程度,以期改善预后[168-169]。非药物治疗,必须是戒烟,对每一位COPD患者都是必不可少的,并且是最有效和最划算的治疗策略,戒烟可以减缓肺功能的丧失,以提高生存率[168-169]。其他非药物治疗方法,包括有规律的体育活动、充足的营养状况、共病的管理以及对所有的患者接种肺炎球菌和流感病毒疫苗,以降低COPD患者急性加重的发病风险,这对于轻中度COPD患者可以从短期和长期康复中获益[170-172]。药物治疗的目的主要是缓解症状和减轻COPD患者临床表现来提高生活质量,降低患者未来健康恶化的风险[173]。可采用短效β2-受体激动剂(short-acting β2-agonists, SABA),对于SABA难以控制的患者,可选择长效胆碱能抗剂(long-acting anti-muscarinic antagonists, LAMA)、长效β2-受体激动剂(long-acting β2-agonists, LABA)、ICS联合用。甲基黄嘌呤类药物单用或与SABA联合使用,沙丁醇胺单用或与异丙托溴胺联合常用于急救治疗。LABA和LAMA虽然通过不同的作用机制扩张支气管,都用于维持治疗,通常终生使用[174]。如每天吸入1次的LAMA制剂噻托溴胺,或每天吸入2次的LABA制剂沙美特罗和福莫特罗,其中LAMA比LABA的支气管扩张作用更优[169]。
目前的观察,对COPD患者初治者联合应用乌米昔丁(umeclidinium)和维兰特罗(vilanterol)(LAMA/LABA)与替奥妥平(tiotropium)比较,结果表明,早期使用双支气管扩张剂治疗COPD伴有症状者,对肺功能改善具有较好的疗效,与单药治疗相比,可降低短期临床严重恶化(clinically important deterioration, CID)的风险[175]。关于阿昔丁胺(aclidinium)/福 莫特罗(formoterol)联合治疗COPD患者的疗效和安全性比较,结果表明,与安慰剂或单一药物疗效相比,这种组合降低了首次CID的风险,提供了更大的气道稳定性,因此,肺功能恶化、呼吸困难、恶化风险和健康状况恶化的情况更少[176]。一篇关于COPD治疗指南指出,尽管在一些推荐的治疗方法上存在差异,但在治疗目标和长效支气管扩张剂(long-acting bronchodilators, LABD)作为COPD治疗的基础上存在普遍共识[177]。所以,对于症状稳定、无频繁加重的COPD患者,应以LABD开始治疗,如果出现持续或加重的呼吸困难、运动不耐受和/或使用单药治疗后健康状况恶化,应考虑接受LABA/ICS或LAMA/LABA加强治疗。对于稳定的COPD患者,尽管使用LAMA或LABA单药治疗,但COPD病情仍在恶化,应考虑加强吸入LAMA/LABA治疗。如果患者在使用LAMA/LABA的情况下病情仍在恶化,可以考虑使用LAMA+LABC/ICS加强治疗。由于吸入三联疗法或双联疗法不一定对每例COPD患者都有疗效,因此,应考虑患者其他问题。对于疑似ACOS的患者,临床上推荐LABA/ICS联合使用[178]。中重度COPD患者的治疗流程,见图5。

图5 GOLDⅠ-Ⅱ COPD患者管理治疗流程建议;注:COPD:慢性阻塞性肺疾病;ACOS或ACO:哮喘-COPD重叠综合征;LAMA:长效胆碱能拮抗剂;LABA:长效β-2受体激动剂;ICS:吸入型皮质类固醇;GOLD:全球慢性阻塞性肺疾病防治创议;mMRC dyspnea:呼吸困难量表
由于COPD是进行性发展的,关键是在早期识别和治疗COPD患者,以防止病情进一步恶化至关重要,更重要的是考虑到轻度COPD患者已经有测量到的生理损害,导致生活方式的改变和脏器损伤。早期阶段,通过适当的药物和非药物(如戒烟、生活管理、生活受限和防止病情恶化)的治疗,可积极改善疾病的进展。这些益处在病情较轻的患者中可能比病情较严重的COPD患者更为明显。对于中重度COPD患者,药物治疗效果不佳者,并且病情进一步恶化的COPD患者,如有条件,可进行LVRS或进行肺移植(lung transplantion)[174]。总之,最新的研究表明,对于COPD常规药物治疗,学者们和指南均推荐早期使用双支气管扩张剂治疗可以防止病情恶化[175,177,179-180],以及个体化和精准化治疗。虽然COPD基本病因繁多,发病机制非常复杂,临床治疗十分困惑。目前还没有一种能逆转COPD患者病情进展或治愈的药物,随着科学技术的发展,医学科学的进步,但终究会有一天研发出能治愈COPD患者的新的药物或疗法。希望医学科学家共同努力,齐心协力,方能为COPD患者创造美好的将来,最终控制COPD这种灾难性疾病。
猜你喜欢
保健医苑(2022年1期)2022-08-30
中老年保健(2021年11期)2021-08-22
现代临床医学(2021年4期)2021-07-31
昆明医科大学学报(2021年3期)2021-07-22
小学阅读指南·低年级版(2021年3期)2021-03-19
文史博览(2020年1期)2020-11-30
爱你(2020年22期)2020-11-19
文史博览·文史(2020年1期)2020-03-12
华人时刊(2019年13期)2019-11-26
小猕猴学习画刊(2018年10期)2018-11-23
